


ISSN: 0973-7510
E-ISSN: 2581-690X
MicroRNAs (miRNAs) are small non-encoding RNAs of 19-24 nucleotides long. It regulates gene expression through target mRNA degradation or translational gene silencing. Experimental based prediction is laborious and economically unfavorable due to a huge number of miRNAs and potential targets. So researchers are focused on computational approach for faster prediction. A large number of computational based prediction tools have been developed, but their results are often inconsistent. Hence, finding a reliable computational based prediction tool is still a challenging task. Here we proposed a computational method, microTarget for finding miRNA – mRNA target interactions. We validated our result in C. elegans and Rattus norvegicus genomes and compared performance with three computational methods, like miRanda, PITA, and RNAhybrid. Signal-to-noise ratio, z score, Receiver operating characteristic (ROC) curve analysis, Matthews correlation coefficient (MCC) and F measure show that microTarget exhibits good performance than other three miRNA – mRNA target interactions methods used in this study.
miRNA – mRNA target interactions, Target validation, Complementarity score, Computational methods.
miRNAs are first identified in the year 1993 using genetic methods1 in Caenorhabditis elegans. miRNAs are small, non-coding, endogenous RNAs that can negatively control their target gene expression post-transcriptionally2 and perform an important regulator of gene expression in many biological systems. miRNAs are expressed from long transcripts produced in animals, plants, viruses, and single-celled eukaryotes3. miRNAs have become the focus of many researchers because of their significant role in the degradation of mRNA, post-translational inhibition through complementary base pairing4, and ability to control many biological processes such as homeostasis3. miRNA regulates the target mRNA to make adjustments to the forming corresponding protein, which dysregulates the functions of miRNA, thereby leading to several human diseases like cancer, viral infection etc.5, 6, 7, 8, 9. A large amount of miRNA data has been generated in recent years. Due to the major efforts in identifying their targets and functions, a computational method is preferable than biological methods as it provides statistical approaches to assess their quality and accuracy. Some features used by computational approaches for the mammalian target prediction programs are based on base pairing pattern, thermodynamic stability, comparative sequence analysis, the presence of multiple target sites. Some widely used miRNA target prediction algorithms are miRanda10, PITA11, RNAhybrid12 etc. RNAhybrid and PITA are based on thermodynamics. RNAhybrid computes scores based on secondary structure, whereas PITA assesses the accessibility of the site (seed match) by the difference between the minimum free energy of the duplex and the energy required to unpair and open the target site. miRanda is based on three features: comparison of miRNA complementarity of 3′ UTR regions, free energies of RNA-RNA duplexes, and conservation of target sites in related genomes, but due to the conservation of target sites, it can’t be used universally. The accuracy of miRNA target prediction can be improved with the use of positive and perfect negative set. Positive examples can be obtained from the available experimentally verified miRNA target databases such as MirTarBase database 13. In the earlier machine learning approaches, randomly generated sequences were used as negative examples. However, such sequences often interact with miRNAs, as shown in the signal-to-noise ratio experiments of previous studies14, 15. miRanda10 was then used to predict the targets of a randomly chosen subset of 100 such artificial miRNA. These artificial miRNA–target pairs were used as the negative data. These randomly generated negative examples may contain real cases by chance. To avoid these cases negative data are generated using mock miRNAs, in a manner similar to the approaches used in John et al.16 and Maragkakis et al.17. To improve false positive rate in our algorithm we have incorporated the results of Brennecke et al18 and Xiaowei and Wang19, and Grimson et al.20 in our algorithm. In this article, we have proposed our new algorithm microTarget and tried to validate in C. elegans and Rattus norvegicus genomes. We have validated microTarget with experimental results and compare validation results with miRanda, PITA, and RNAhybid. Statistical measures like signal-to-noise ratio, z score, MCC score, F-measure and ROC curve are calculated and compared results with miRanda, PITA, and RNAhybid in C. elegans and Rattus norvegicus genomes.
Positive data
We considered experimentally validated data obtained from miRTarBase database13 as positive data set. 1542 experimentally validated miRNA- gene pairs of C. elegans genome and 387 miRNA-gene pairs of Rattus norvegicus genome are obtained from miRTarBase and used as a positive set. We have also downloaded 3′ UTR of target genes of C. elegans genome and Rattus norvegicus genome from UTRdb21.
Negative data
The negative data set are produced using mock miRNAs in the procedure described in John et al.16 and Maragkakis et al17. Mock miRNAs are produced by random rearrangement of an actual miRNA sequence in such a way that mock miRNA and actual miRNA don’t show any similarity in seed region. Every actual miRNA is permuted randomly using Fisher-Yates shuffle algorithm22 until 7mer seed sequence of permuted miRNA does not coincide with 7mer of the seed sequence of every actual miRNAs enlisted in miRTarBase database, and then we call it a mock miRNA. Mock miRNA-gene pairs are made for every actual miRNA-3’ UTR of the positive dataset. We have used 113 miRNA sequences and 305 3’UTR sequences for C. elegans genome and 113 miRNA sequences and 153 3’UTR sequences for Rattus norvegicus genome as the negative set.
microTarget algorithm
microTarget algorithm23 is similar to the miRanda algorithm (Enright et al. 2004), however, instead of using empirical rules. It uses similar complementarity parameters as miRanda algorithm uses at every aligned position: +5 for GºC, +5 for A=U, +2 for G=U and -3 for all other nucleotide pairs. The algorithm uses affine penalties for gap-opening (-8) and gap-extension (-1). Also, the scores of the first 11 positions from the 5′ end of the miRNA are multiplied by 2. The following five rules apply to the positions from 5′ end of the miRNA:
(1) There must be 6 to 8 base pairs between positions 1 to 10.
(2) Seed region with 8 base pairs and starting from position 1, may have up to two G=U base-pairs or one bulge (either of the miRNA or of the 3’UTR) or single non-G=U mismatch in between the seed region (i.e. from positions 2-7).
(3) Seed region with 7 base pairs and starting from positions 1-4, may have one G=U base-pair or one bulge (either of the miRNA or of the 3’UTR) or single non-G=U mismatch in between seed region.
(4) Seed region with 6 base pairs and starting from positions 2-5, may have only one G=U base-pair in between seed region.
(5) If G=U base pair or bulge or mismatch is used in seed region and starting from positions either 3-4 or 4-5, there must be at least 4 base pairs (including G=U base-pairs) from positions 12 to 3’ UTR end of miRNA.
Complementarity score of a miRNA -3’ UTR pair is calculated using the parameters and rules mentioned above and optimized using dynamic programming and then summed over all aligned positions. This miRNA and 3’ UTR interaction will be called as a possible target if its complementarity score is greater than 80 (default value). All the non-overlapping hybridization alignments in decreasing order of complementarity score are also found. In order to calculate free energies of the RNA: RNA duplexes, we use folding routines from the Vienna RNA secondary structure programming library (RNAlib)24. The thresholds used for the possible target are complementarity score > 80 and the energy of the duplex structure < -10 kcal/Mol. All possible miRNA-3’ UTR interaction sites are ranked according to their highest total score and lowest total energy. Only the top 10 ranked miRNA-3’UTR interaction sites are selected as its candidate target genes for each miRNA. A target gene if tied with multiple miRNAs, is selected by the miRNAs for which it scores highest score and lowest free energy so that same miRNA-3’ UTR site is predicted by more than one miRNAs.
Randomized test
We performed similar randomized test as mentioned in Enright et al10. Each randomized miRNA was constructed by retaining its base composition of nucleotides and changing the position of nucleotides taking random one at a time. 100 sets of all miRNAs for each genome of C. elegans and Rattus norvegicus were used in this study. Each of 100 sets of randomized miRNAs was individually investigated against all 3’UTR of target genes for each genome of C. elegans and Rattus norvegicus downloaded from UTRdb. Actual miRNA counts and counts averaged over all 100 random sets and their standard deviations were used to calculate Z-scores for each genome of C. elegans and Rattus norvegicus.
Validation of results of miRanda, PITA, RNAhybrid and microTarget
In this study, 1542 experimentally validated miRNA- gene pairs of C. elegans genome and 387 miRNA-gene pairs of Rattus norvegicus genome are used as a positive set and we have selected three other widely used algortihms, namely miRanda, PITA and RNAhybrid in addition to our algorithm microTarget. The newest versions of miRanda (microrna.org; Enright et al.10), PITA11 and RNAhybrid12 executables were taken and executed with its default parameters as described in the package.
Table 1 shows the number of miRNA-gene intercations by miRanda, PITA, RNAhybrid and microTarget in C. elegans and Rattus norvegicus genomes. PITA predicts the highest number of miRNA-target interactions in the positive and negative set, whereas microTarget predicts a good number of miRNA-target interactions in positive set and less number of interactions in negative set. RNAhybrid predicts very less number of miRNA-target interactions in both positive and negative set. PITA showed a high number of miRNA-target interactions and RNAhybrid showed less number of miRNA-target interactions due their sensitivity and specificity.
Table (1):
Number of miRNA-target interactions by different algorithms in C. elegans and Rattus norvegicus genomes.
| C. elegans | Rattus norvegicus | |||
|---|---|---|---|---|
| No. of miRNA-target interactions in positive set | No. of miRNA-target interactions in negative set | No. of miRNA-target interactions in positive set | No. of miRNA-target interactions in negative set | |
| miRanda | 474 | 285 | 222 | 142 |
| PITA | 1121 | 658 | 291 | 221 |
| RNAhybrid | 194 | 112 | 57 | 22 |
| microTarget | 950 | 265 | 243 | 131 |
Comparing performance of microTarget at the target level
In this study, we have chosen four features- T’s frequency in effective seed matching sight, G: T in seed region, number of G: T matches in the total region and free energy in the seed region as all those features are common in all three target prediction algorithms. The discriminating power of each individual feature is assessed as the marginal distribution in the histogram in both positive and negative set. Figure 1 and 2 show the histogram of selected features in C. elegans and Rattus norvegicus genomes respectively.
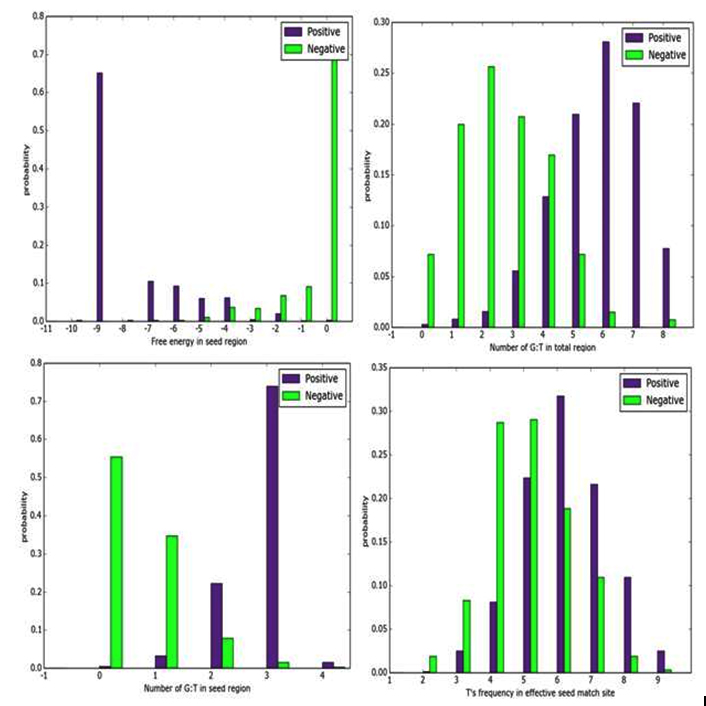
Fig. 1. Histogram of selected features in C. elegans genome
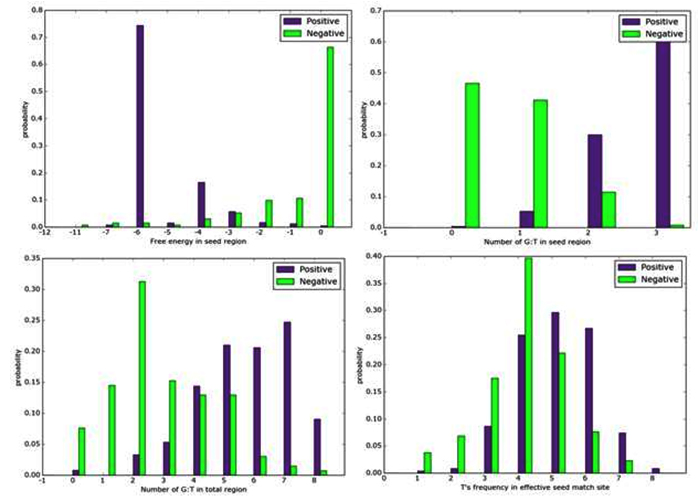
Fig. 2. Histogram of selected features in R. norvegicus genome
We have also assessed the performance of microTarget with other algorithms in terms of sensitivity, Sn=(TP/TP+FN), specificity Sp=(TN/TN+FP), Matthew’s correlation coefficient (MCC) MCC=(TP×TN-FP×FN /√((TP+FP )×(TN+FN )×(TP+FN)×(TN+FP))) and F-measure=(2TP/2TP+FP+FN) where TP = true positive, TN= true negative, FN = false negative and FP = false positive. In this paper, we have evaluated the Receiver Operating Characteristic (ROC) performance of miRanda, RNAhybrid, PITA and microTarget algorithm. ROC performance is normally evaluated as a plot of sensitivity vs. 1- specificity. Figure 3 and 4 show the ROC curves of four miRNA- target prediction algorithms in C. elegans and Rattus norvegicus genomes respectively. In C. elegans, AUC (area under the curve) of microTarget is 0.89, whereas AUC of miRanda, PITA and RNAhybrid are 0.82, 0.77 and 0.78 respectively. In Rattus norvegicus, AUC of microTarget is 0.86, whereas AUC of miRanda, PITA and RNAhybrid are 0.75, 0.75 and 0.76 respectively. It is clear that microTarget performs well than other three miRNA target prediction algorithms.
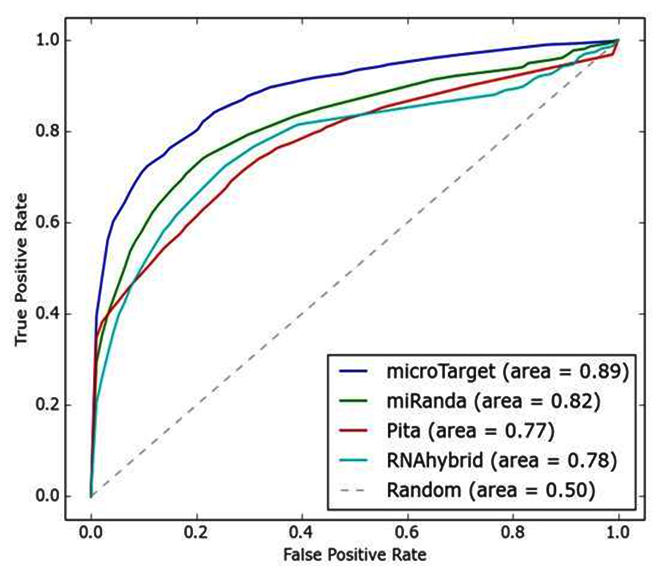
Fig. 3. The ROC curves of different algorithms in C. elegans genome (AUC is shown in the bracket)
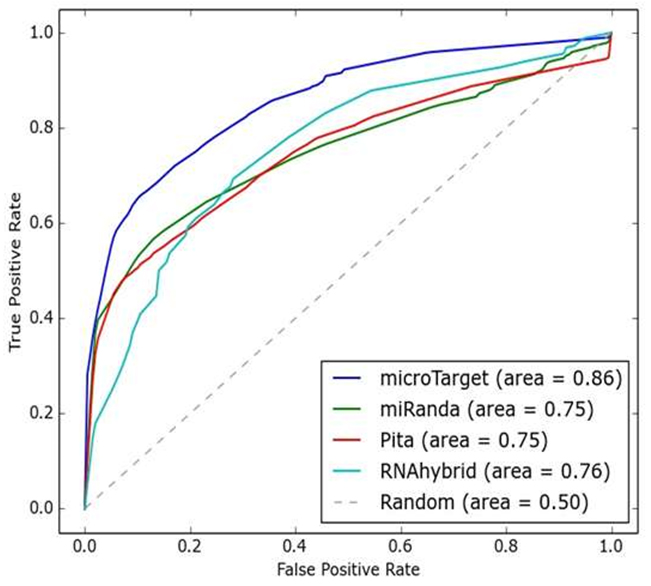
Fig. 4. The ROC curves of different algorithms in R. norvegicus genome (AUC is shown in the bracket)
Table 2 shows the MCC score in both the genomes of C. elegans and Rattus norvegicus. MCC scores of microTarget in C. elegans and Rattus norvegicus are 0.45 and 0.29 respectively, but MCC scores of other three algorithms are less than 0.21 in both genomes. Table 3 shows the F -measure in both the genomes of C. elegans and Rattus norvegicus. microTarget shows 0.69 and 0.6 F-measure in C. elegans and Rattus norvegicus respectively, but F-measure of other three algorithms are less than microTarget in both genomes. It can be easily verified that, at any fixed true positive rate (TPR), microTarget provides the lowest false positive rate (FPR) and at the same time, for any fixed FPR, the TPR of microTarget is higher than those of all the three target prediction algorithms.
Table (2):
MCC scores of four algorithms in C. elegans and Rattus norvegicus genomes.
| MCC Score | ||||
|---|---|---|---|---|
| miRanda | PITA | RNAhybrid | microTarget | |
| C. elegans | 0.21 | 0.30 | 0.09 | 0.45 |
| Rattus norvegicus | 0.21 | 0.15 | 0.15 | 0.29 |
Table (3):
F-measures of four algorithms in C. elegans and Rattus norvegicus genomes.
| F-measure | ||||
|---|---|---|---|---|
| miRanda | PITA | RNAhybrid | microTarget | |
| C. elegans | 0.41 | 0.67 | 0.21 | 0.69 |
| Rattus norvegicus | 0.59 | 0.63 | 0.24 | 0.64 |
Analysis of signal-noise ratio of all four algorithms
The signal-to-noise ratio is another way of validating results. It is demarcated as the ratio between average no. of predicted targets by actual miRNA and the average number. of predicted targets by randomized miRNA in searched 3’UTR. Table 4 shows the signal-to-noise ratio of miRanda, RNAhybrid, PITA, and microTarget in two genomes. It is clear from Table 4 that signal-to-noise ratio of microTarget is greater than 2 in both the genomes. We have also calculated the z score of miRanda, RNAhybrid, PITA and microTarget in two genomes (shown in Table 4) and microTarget showed highest z-score in C. elegans and Rattus norvegicus genomes. These results indicate that microTarget is significantly predicted miRNA-target interactions than miRanda, RNAhybrid, and PITA.
Table (4):
Signal-to-noise ratios and Z-scores of miRanda, PITA, RNAhybrid and microTarget in C. elegans and Rattus norvegicus genomes.
| C. elegans | Rattus norvegicus | |||||||
|---|---|---|---|---|---|---|---|---|
| miRanda | PITA | RNAhybrid | microTarget | miRanda | PITA | RNAhybrid | microTarget | |
| Average no. of predicted targer gene per miRNA | 4.35 | 11.10 | 1.78 | 5.78 | 1.96 | 2.58 | 0.50 | 2.15 |
| Average no. of predicted targer gene per randomized miRNA | 2.74 | 5.98 | 0.92 | 2.44 | 1.22 | 1.97 | 0.26 | 1.04 |
| Standard Deviation | 0.13 | 0.20 | 0.08 | 0.13 | 0.08 | 0.06 | 0.04 | 0.06 |
| Signal-to-noise ratio | 1.59 | 1.86 | 1.93 | 2.37 | 1.61 | 1.31 | 1.92 | 2.07 |
| Z Score | 12.80 | 25.64 | 10.15 | 26.12 | 9.03 | 9.53 | 6.67 | 17.28 |
In this article, we have proposed our new algorithm microTarget and tried to validate in C. elegans and Rattus norvegicus genomes. Experimentally validated results downloaded from MirTarBase database of C. elegans and Rattus norvegicus genomes are used as positive set and results showed that microTarget performs better than other three target prediction methods. Statistical measures like a signal-to-noise ratio, z score, MCC score, F-measure and ROC curve are calculated and results showed that performance of mocroTarget is quite satisfactory than miRanda, PITA, and RNAhybrid.
- Lee, R. C., Feinbaum, R. L., and Ambros, V. The C. elegansheterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell, 1993,75(5), 843-854.
- Chuang, J.; Jones, P. Epigenetics and MicroRNAs. Pediatr. Res. 2007, 61: 24R–29R.
- Liu, B., Li, J., & Cairns, M. J. Identifying miRNAs, targets and functions. Briefings in bioinformatics, 2012, 15(1): 1-19.
- He, L., and Hannon, G. J. MicroRNAs: small RNAs with a big role in gene regulation. Nature Reviews Genetics, 2004, 5(7): 522-531
- Yanaihara, N., Caplen, N., Bowman, E., Seike, M., Kumamoto, K., Yi, M., and Calin, G. A. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer cell, 2006, 9(3): 189-198.
- Porkka, K. P., Pfeiffer, M. J., Waltering, K. K., Vessella, R. L., Tammela, T. L., and Visakorpi, T. MicroRNA expression profiling in prostate cancer. Cancer research, 2007, 67(13), 6130-6135.
- Yang, H., Kong, W., He, L., Zhao, J. J., O’Donnell, J. D., Wang, J., and Cheng, J. Q. MicroRNA expression profiling in human ovarian cancer: miR-214 induces cell survival and cisplatin resistance by targeting PTEN. Cancer research, 2008, 68(2): 425-433.
- Beveridge, N. J., Gardiner, E., Carroll, A. P., Tooney, P. A., and Cairns, M. J. Schizophrenia is associated with an increase in cortical microRNA biogenesis. Molecular psychiatry, 2010, 15(12): 1176-1189.
- Cox, M. B., Cairns, M. J., Gandhi, K. S., Carroll, A. P., Moscovis, S., Stewart, G. J., and ANZgene Multiple Sclerosis Genetics Consortium. MicroRNAs miR-17 and miR-20a inhibit T cell activation genes and are under-expressed in MS whole blood. PloS one, 2010, 5(8): e12132.
- Enright A.J., John B., Gaul U., Tuschl T., Sander C. and Marks DS. MicroRNA targets in Drosophila. Genome Biol., 2004; 5(1): R1-14.
- Kertesz,M. et al. The role of site accessibility in microRNA target recognition. Nat. Genet., 2007, 39: 1278–1284.
- Rehmsmeier, M.,Steffen, P., Hochsmann, M. and Giegerich, R. Fast and effective prediction of microRNA/target duplexes. RNA, 2004, 10(10), 1507-1517.
- Chou CH, Chang NW, Shrestha S, Hsu SD, Lin YL, Lee WH, Yang CD, Hong HC, Wei TY, Tu SJ, Tsai TR, Ho SY, Jian TY, Wu HY, Chen PR, Lin NC, Huang HT, Yang TL, Pai CY, Tai CS, Chen WL, Huang CY, Liu CC, Weng SL, Liao KW, Hsu WL and Huang HD. miRTarBase 2016: updates to the experimentally validated miRNA-target interactions database. Nucleic Acids Res. 2016; 44 (D1):239-47
- Krek,A. et al. Combinatorial microRNA target predictions. Nat. Genet., 2005; 37: 495–500.
- Lewis,B.P. et al. Prediction of mammalian microRNA targets. Cell, 2003; 115: 787–798.
- John B., Enright A.J., Aravin A., Tuschl T.., Sander C and Marks D.S.: Human microRNA targets. PLoS Biol 2004, 2: e363.
- Maragkakis M., Alexiou P., Papadopoulos GL., Reczko M., Dalamagas T., Giannopoulos G., Goumas G., Koukis E., Kourtis K. and Simossis V.A.: Accurate microRNA target prediction correlates with protein repression levels. BMC Bioinformatics 2009; 10: 295.
- Brennecke J., Stark A., Russell R.B. and Cohen S.M. Principles of microRNA–target recognition. PLoS Biol, 2005; 3(3): e85.
- Xiaowei Wang. Composition of seed sequence is a major determinant of microRNA targeting patterns. Bioinformatics, 2014; 30 (10):1377–1383.
- Grimson A., Farh K.K., Johnston W.K., Garrett-Engele P., Lim L.P. and Bartel D.P. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell. , 2007; 27(1): 91-105.
- Giorgio G., Antonio T., Flavio, L., Flavio M., Sabino L., Sandro B., Vincenzo A.G., David S.H.,Giulio P., Ernesto P. and Graziano P. UTRdb and UTRsite (RELEASE 2010): a collection of sequences and regulatory motifs of the untranslated regions of eukaryotic mRNAs. Nucleic Acids Res. 2010; 38 (Database issue): D75–D80.
- Knuth D.E: The Art of Computer Programming: Seminumerical Algorithms II, Volume 2. Boston, MA: Addison-Wesley; 2014.
- Das S. and Mandal D. MicroRNA Target Gene Prediction and Validation in Different Genomes. Rsearch Journal of Life Science, Bioinformatics, Pharmaceutical and Chemical Science, 2016; 2(2) 10 – 28.
- Wuchty S., Fontana W., Hofacker I.L. and Schuster P. Complete suboptimal folding of RNA and the stability of secondary structures. Biopolymers, 1999; 49: 145-165.
© The Author(s) 2018. Open Access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License which permits unrestricted use, sharing, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.