


ISSN: 0973-7510
E-ISSN: 2581-690X
In this study, considering the vital biological importance of the Alongshan virus NS3-like helicase enzyme, several machine learning and artificial intelligence-based software and servers were used to identify compounds that exhibited the best binding affinity for the helicase enzyme. The predicted compounds were MSID000152, MSID000165, MSID000200, AfroDb.28, and AfroDb.207 with binding energy scores of -9.7, -9.5, -9.4, -8.65, and -8.01 kcal/mol, respectively. Because static intermolecular confirmation is not highly valued in terms of docked stability, the results were validated through molecular dynamics simulation analysis within 100 ns. The MSID000152, MSID000165, and MSID000200 showed significant uniform dynamics with root mean square deviation (RMSD) values of <3 Å. The intermolecular interaction energies were estimated using two well-known methods: MMPBSA and WaterSwap. Both methods agreed regarding the appreciated intermolecular strength of the leads with the helicase enzyme. Van der Waals interactions were identified to be the dominant force in stabilizing the ligands with the helicase enzyme in all complexes. Similarly, the electrostatic energy supported the stable intermolecular conformation of the docked complexes. The selected compounds were drug-like and exhibited good pharmacokinetic properties.
Alongshan Virus, NS3-Like Helicase Enzyme, Structure Based Virtual Screening, AI, Molecular Dynamics Simulation
Viral infections cause millions of deaths worldwide. The Alongshan virus is a single-stranded RNA virus belonging to the Flaviviridae family that has been identified in tick-biting patients and Ixodes persulcatus in northeast China.1 Subsequently, the Alongshan virus was identified in sheep and cattle in endemic regions. The clinical manifestation of the Alongshan virus is reminiscent of those of other tick-borne viruses that cause nonspecific febrile diseases. Notably, Alongshan fever, caused by Alongshan viral infection, is characterized by several clinical symptoms, including fever, headache, coma, exhaustion, depression, nausea, myalgia, arthralgia, and rash.2 Its genome has been segmented and classified within the Jingmenvirus family of Flaviviridae. The virus is tagged as an emerging virus, and its spread across countries poses potential threats to human health and healthcare systems; therefore, a detailed study of the genome-encoded proteins of this virus from a therapeutic perspective is vital.3
The genome of the Alongshan virus was divided into four segments, designated as segments 1-4. The total genome size is similar to that of other flaviviruses.4 Segment 1 encodes the flavivirus NS5 protein, which comprises an RNA-dependent RNA polymerase enzyme and methyltransferase motifs, whereas segment 2 encodes the product of the NS2b-NS3 complex, which contains an RNA helicase motif and proteinase.5 Segments 2 and 4 are expressed as VP1-3 proteins. The NS3 protein of flaviviruses is among the most studied nonstructural proteins because it plays a vital role in viral replication.6 This enzyme is a key component of the replication complex in the membrane and exhibits various enzymatic activities.7 The enzyme contains an N-terminal domain (designated as the protease domain) and a C-terminal domain (designated as the RNA helicase domain). The proteinase domain is involved in polyprotein processing, whereas the RNA helicase domain is involved in RNA synthesis and capping.5 The NS3 helicase enzymes from different viruses have been reported to have a similar structural fold, a similar mechanism regarding RNA recognition and unwinding, and adenosine triphosphate (ATP) hydrolysis. Owing to the key involvement of the NS3 helicase enzyme in the biology of the Alongshan virus, it is a promising target of small drug molecules that may block its enzymatic function.8
Computer-aided drug design (CADD) is an integral part of modern drug discovery for predicting target-ligand interactions and is essential in understanding drug safety for therapeutic use.9 CADD allows the screening of tens of thousands of compounds to identify molecules with special effects on the target enzyme. Over the years, CADD technology has progressed significantly and has been divided into ligand-based drug design (LBDD) and structure-based drug design (SBDD).10 The integration of artificial intelligence and machine learning with CADD has recently attracted considerable attention for accomplishing specific tasks.11 AI algorithms are widely used in CADD to predict protein three-dimensional (3D) structures, docking scoring functions, de novo drug design, prediction of synthetic accessibility, and synthetic routes.12 Several drugs, including captopril, oseltamivir, dorzolamide, rupintrivir, and zanamivir, have been successfully developed using CADD.13 In the present study, several CADD techniques based on machine learning and AI principles were used to identify drug molecules from natural sources that showed stable binding conformations with the NS3 helicase enzyme of the Alongshan virus. The virtual screening process identified the best-docked complexes, which were then validated through molecular dynamics simulation analysis.14 MD simulations allowed us to decipher complex time-dependent dynamics and investigate ligand-binding stability. Furthermore, the intermolecular binding free energies were determined using different approaches to determine the dominant interaction energies.15,16 Finally, the drug-likeness and pharmacokinetic properties of the selected compounds were evaluated to improve the clinical and developmental efficacy of the drugs and reduce production time and costs.17
The overall flow of methods used to identify drug molecules targeting NS3-like helicase enzymes is shown in Figure 1.
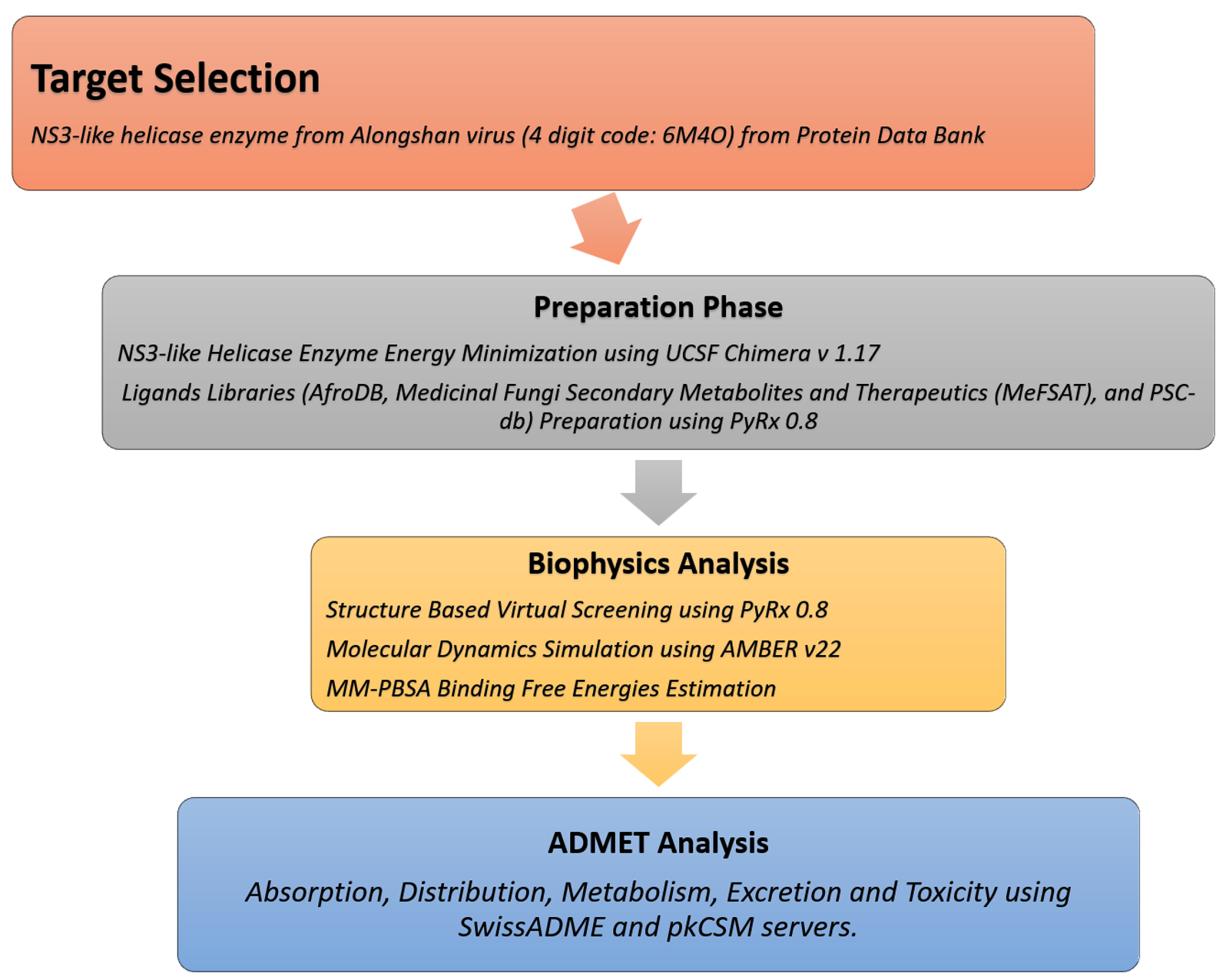
Figure 1. Flow of the designed materials and methods sections employed to achieve the study objectives by identifying drug molecules against NS3-like helicase enzyme from Alongshan virus. The flow is divided into four phases i.e. target selection of NS3-like helicase enzyme from Alongshan virus, preparation phase involving preparation of target and drug libraries, biophysics phase which involves structure-based virtual screening, molecular dynamics simulation and MM-PBSA analysis, and ADMET analysis phase
NS3-like helicase structure preparation
The crystal structure of the NS3-like helicase enzyme of the Alongshan virus was downloaded from the Protein Data Bank (PDB) (https://www.rcsb.org/).18 This etching step was completed by entering a four-digit code, “6M4O”, in the search box of the PDB database.5 The selected structure was expressed in the Escherichia coli BL21 strain and no mutations were reported. The structure was deposited in the PDB database on 05-03-2020 and released on 08-04-2020. The structure was resolved using X-ray diffraction (XRD) at a resolution value of 2.89 Å. The structure has a one-ampere chain and a sequence length of 489 bp. The enzyme structure entered the preparation phase, where it was energy-minimized using the steepest descent and conjugate gradient algorithms.19 Both algorithms were applied to the structure in 1000 steps. During this process, missing hydrogen atoms were added and charges were assigned using the Gasteiger method. After this process, the obtained structure was saved in PDB format.
Ligands library preparation
Different drug libraries were used in this study, including AfroDB,20 Medicinal Fungi Secondary Metabolites and Therapeutics (MeFSAT),21 and PSC-db.22 AfroDB (https://zinc12.docking.org/pbcs/afronp) is a structurally diverse database containing >1000 compounds from African medicinal plants that have been shown to exhibit a range of biological activities. In addition, the catalog of the ZINC database includes natural products originating from various geographic regions in Africa. MeFSAT (https://cb.imsc.res.in/mefsat/) comprises secondary metabolites from 184 medicinal fungi. The Plant Secondary Compound Database (PSCDB;) contains 2853 PSCs that can be screened for bioactivity against various disease targets.22 The database classifies all the compounds into nine subfamilies: (1) polyketides, (2) phenylpropanoids, (3) flavonoids, (4) amino acid-related compounds, (5) acetate-malonate pathway (6) alkaloids, (7) terpenoids, (8) fatty acid-based compounds and (9) others. All library compounds were imported into PyRx 0.8, where they were energy-minimized using an MM2 force field.23 The compounds were then converted to the PDBQT format.
Virtual screening process
Virtual screening is an important foundation in any drug discovery process as it identifies potential therapeutic compounds that can be used as novel inhibitors of the pathophysiology of the disease. The structure-based virtual screening of the drug libraries described above was performed against the fully active pockets available on the surface of the NS3-like helicase enzyme using the PyRx 0.8 software.24 During the screening process, the grid box was set on the receptor enzyme at the X-axes, Y-axes, and Z-axes with dimensions values of 38.79, 38.57, and 73.40 Å, respectively. The box size in each direction was adjusted to 49.40, 62.20, and 67.21 Å, respectively. Fifty binding conformations were obtained for each ligand during the virtual screening. Furthermore, selective side-chain residue flexibility can increase the accuracy of the Vina docking score without appreciably increasing the processing time. Therefore, AutoDock Vina uses the selective side-chain residue flexibility option, which provides a more realistic ligand-protein interaction environment without significantly increasing the computer processing time.25 The lowest energy (kcal/mol) binding modes were filtered as the best binding conformation and selected for visualization. In addition, 6-(3,5-diaminophenyl)-1-[4-(propan-2-yl)benzyl]-1H-indol-3-yl acetic acid was used as a control. UCSF Chimera 1.17 was used to study the intermolecular binding of the receptor-ligand complexes, whereas Discovery Studio Visualizer 2021 was used to investigate the binding interactions.26,27
MD simulation
Only the top complexes were used in MD simulation analysis to understand the intermolecular binding conformation dynamics of the receptors and ligands.28 AMBER 22 program was used for the analysis.29 Complex files were prepared using Antechamber software. FF19SB was selected as the force field to describe the parameters of the NS3-like helicase enzyme, whereas organic ligands were treated with a gaff 2 force field.30 Energy minimization of the complexes was performed in 1000 steps with applied constraints.31 The complexes were submerged in an oxygen atom carries water box, followed by the addition of 12 Na+ ions.32 The complexes were then heated slowly to 310 K (through Langevin dynamics) while keeping the volume box fixed and equilibrated in the isothermal-isobaric ensemble at 1 atm for 100 ps.33 The production run was completed within 100 ns in the NPT ensemble,34 simulation trajectories were generated and analyzed using the CPPTRAJ module,35 and the plots for different statistical analyses were constructed using XMGRACE v. 5.1.36
Calculation of binding-free energies
The binding free energies of the simulated complexes were determined using the Poisson-Boltzmann surface area (PBSA) method of molecular mechanics (MM) implemented using AMBER v22. This procedure was performed using the MMPBSA.py module.37 PalmTop files for the receptor, ligand, and complex were obtained using the Ante-MMPBSA. py module. The MM-PBSA approach is considered a significant approach in modern drug discovery because of its usefulness in estimating the contribution of different energies to protein-ligand interactions.38 MM-PBSA splits the binding energy into the polar solvation, nonpolar solvation, and vacuum potential energies.39 The solvation energy terms were computed using solvent-accessible surface areas (SASA) and Poisson-Boltzmann equations.
∆G binding = ∆G (vacuum energy) + ∆G (solvation energy)
From the simulation trajectories, 1000 frames were selected across the simulation time and split into equal time intervals.
Normal mode entropy calculations
The entropy energy of each complex was calculated to elucidate the free energy of the complexes further and reveal the real intermolecular binding energy.40 This was achieved using the AMBER normal-mode entropy method, which was applied to selected simulation frames. Five frames were selected from each complex for the entropy analysis.
WaterSwap calculations of absolute binding free energy
The intermolecular binding affinities of the docked complexes were further validated using the WaterSwap absolute binding energy method.41 This method uses Monte Carlo simulation, which facilitates the absolute binding free energy of docked complexes. Furthermore, this procedure calculates the binding energy through free energy perturbation (FEP), thermodynamic integration (TI), and Bennett.42 The consensus of these energies was determined by calculating the arithmetic means.
Prediction of Drug-likeness and absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties
The SwissADME server (http://www.swissadme.ch/), which predicts the drug-like features and pharmacokinetic properties of small selected compounds, was used to predict the drug-likeness of prodigiosin based on several criteria, including the Lipinski rule of five,43 Ghose,44 Egan,45 Veber46 and Muegge rules.47 The SMILES format of prodigiosin was retrieved from the ChEMBL database (https://www.ebi.ac.uk/chembl/) and used as the input.
PkCSM was used to predict the ADMET properties of the selected compounds, as well as their carcinogenicity, solubility, and other pharmacokinetic properties.48,49
Identification of lead compounds
Structure-based virtual screening is an in silico method used in the early drug discovery process to identify novel bioactive molecules in drug libraries against a specific drug biomolecule target.16,50 This method uses a biological receptor target in a 3D structure (obtained through experimental or computational molecular modeling techniques).14 A drug library or several other drug libraries was then docked to the receptor active site, and the binding energy score for each compound in the library was computed.19 Next, a subset of the best-docking compounds was used for biological evaluation. As a result, the process provided selected compounds for experimental testing and thus saves time, resources, and money for the biological assessment of complete drug libraries.34 In this work, the virtual screening process identified the five best-docked compounds with the NS3-like helicase enzyme from the Alongshan virus, which are listed in Table 1.
Table (1):
Selected five compounds considering their lowest binding energy with the NS3-like helicase enzyme
Compound |
Structure |
SMILES |
Binding Energy in kcal/mol |
|---|---|---|---|
MSID000152 |
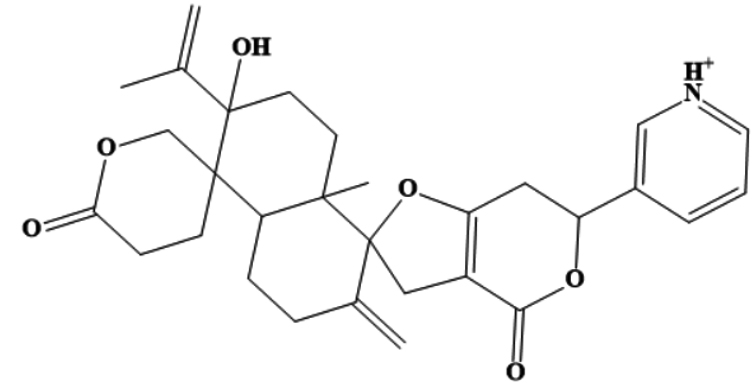 |
CC(=C)C1(O)CCC2(C)C(CCC(=C)C23CC4= C(CC(OC4=O)C5=C[NH+]=CC=C5)O3) C16CCC(=O)OC6 |
-9.7 |
MSID000165 |
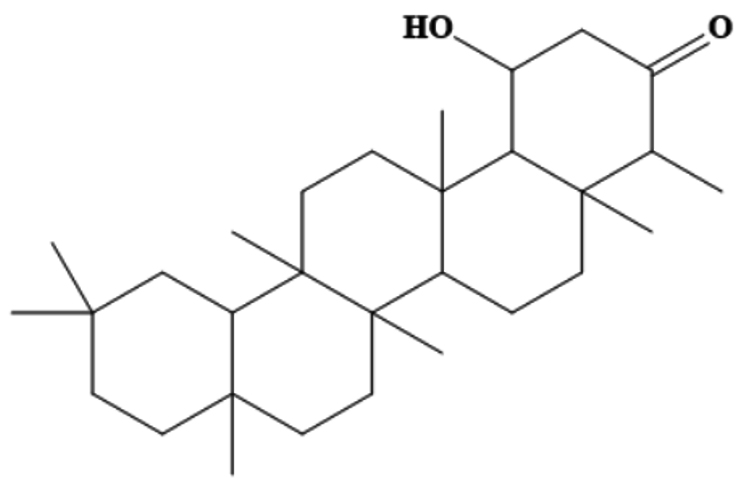 |
CC1C(=O)CC(O)C2C1(C)CCC3C2(C)CCC4(C) C5CC(C)(C)CCC5(C)CCC34C |
-9.5 |
MSID000200 |
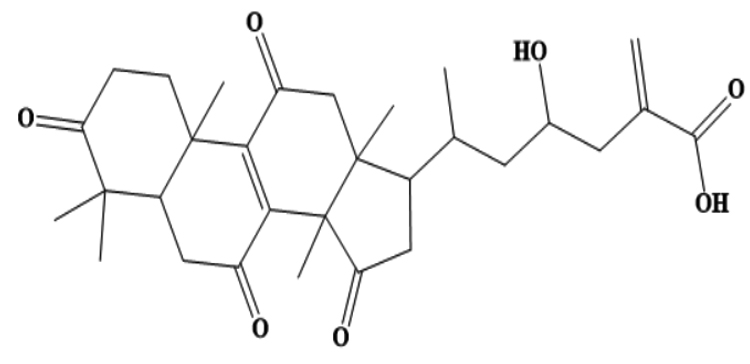 |
CC(CC(O)CC(=C)C(O)=O)C1CC(=O)C2(C)C3=C (C(=O)CC12C)C4(C)CCC(=O)C(C)(C)C4CC3=O |
-9.4 |
AfroDb.28 |
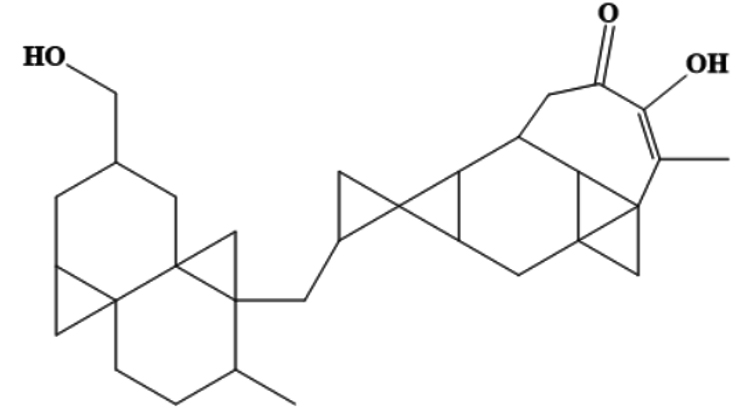 |
CC1CCC23CC2CC(CO)CC34CC14CC5CC56C7CC 89CC8%10C9C(CC(=O)C |
-8.65 |
AfroDb.207 |
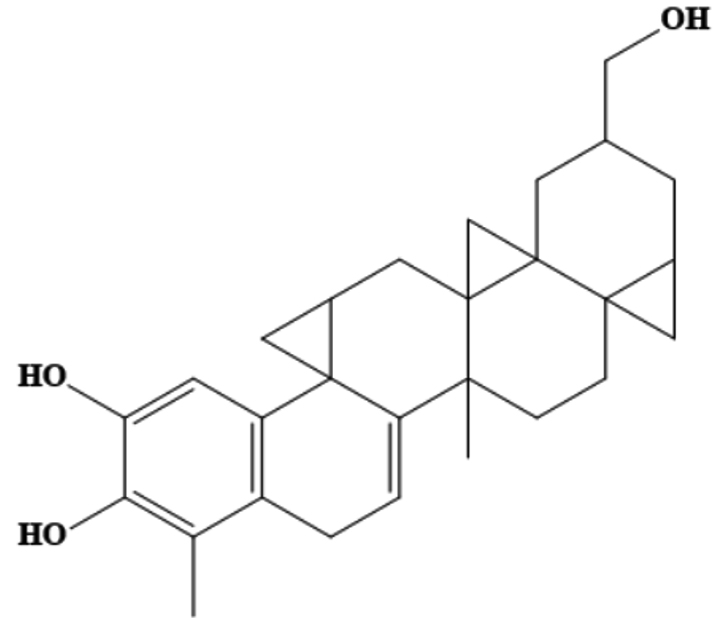 |
CC1=C(O)C(=CC2=C1CC=C3C4(C)CCC56CC5CC (CO)CC67CC47CC8CC238)O |
-8.01 |
Control |
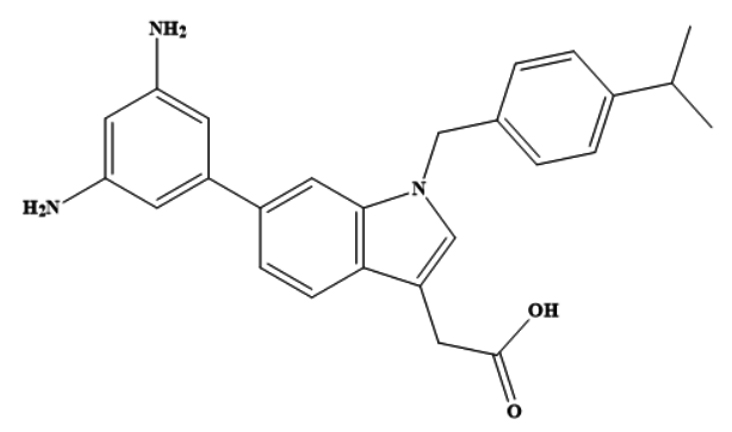 |
6-(3,5-diaminophenyl)-1-[4-(propan-2-yl) benzyl]-1H-indol-3-yl acetic acid |
-8.62 |
These compounds are labeled MSID000152, MSID000165, MSID000200, AfroDb.28, and AfroDb.207 with docking binding energy scores of -9.7, -9.5, -9.4, -8.65, and -8.01 kcal/mol, respectively. All compounds showed preferential binding to the helicase enzyme RNA-binding site, revealing a stable binding-interacting network. Binding of the compounds occurred at the interface of domains D1 and D2. Recent studies have identified compounds that target viral enzymes associated with infectious diseases using computational approaches. Singh et al. showed the potential of ribavirin, levovirin, and ribamidine as effective inhibitors of the tick-borne encephalitis virus, with the lowest docking energies indicating strong binding affinity to the NS3 helicase.50 Ejeh et al. performed structure-based virtual screening to identify novel ketoamide compounds as potential inhibitors of HCV NS3/4A proteases.51 Rehman et al. used a similar in silico approach to identify the natural compounds that target the NS3 protease of the dengue virus and revealed that three compounds, cyanidin 3-glucoside (L22), dithymoquinone (L25), and glabridin (L28), had the strongest binding affinities, with binding energies ranging -8 to -7 kcal/mol.52
Chemically, MSID000152 is 5-(2-(3-hydroxy-4-methylpent-4-en-1-yl)-6-methylene-4’-oxo-3-(6-oxotetrahydro-2H-pyran-3-yl)-3′,4′,6′,7′-tetrahydrospiro [cyclohexane-1,2’-furo[3,2-c]pyran]-6’-yl)-1,2-dihydropyridin-1-ium and forms a network of hydrophobic interactions with the enzyme, especially through its 5-(2-(3-hydroxy-4-methylpent-4-en-1-yl)-6-methylene-4’-oxo-3′,4′,6′,7′-tetrahydrospiro[cyclohexane-1,2’-furo[3,2-c]pyran]-6’-yl)-1,2-dihydropyridin-1-ium chemical region. Hydrophobic interactions were formed with Thr376, Glu576, Val441, Ile574, Ser573, Lys550, Ser548, Thr572, Pro521, Gly522, Thr524, Tyr593, and Gln592. The chemical moiety (tetrahydro-2H-pyran-2-one) of the compound forms a strong hydrogen bond with Leu523 at a distance of 1.67 Å. MSID000165, which is chemically 1-hydroxy-4,4a,6b,8a,11,11,12b,14a-octamethylicosahydropicen-3(2H)-one, was ranked second. The compound formed hydrogen bonds with Gln592 and Pro521 at distances of 1.87 and 2.23 Å, respectively. Hydrophilic interactions form in the chemical region of 5-hydroxy-2,3-dimethylcyclohexanone. The remaining compound (1-ethyl-2,4a,6,6,8a,10a-hexamethyltetradecahydrophenanthrene) formed hydrophobic bonds with Ile574, Arg549, Ser548, Lys550, Ser573, Thr572, Tyr593, Gly522, Leu523, and Thr524. MSID000200 is chemically 4-hydroxy-2-methylene-6-(4,4,10,13, 14-pentamethyl-3,7,11,15-tetraoxo-2,3, 4,5,6,7,10,11,12,13,14,15, 16,17-tetradecahydro-1H-cyclopenta [a]phenanthren-17-yl) heptanoic acid. The methacrylic acid ring of the compound was the most stable in terms of its interaction with the key residues of the enzyme. It produced strong hydrogen bonds with Thr650 and Val649 at distances of 2.36 and 2.97 Å. respectively. Intermolecular docked snapshots of the complexes are shown in Figure 2.
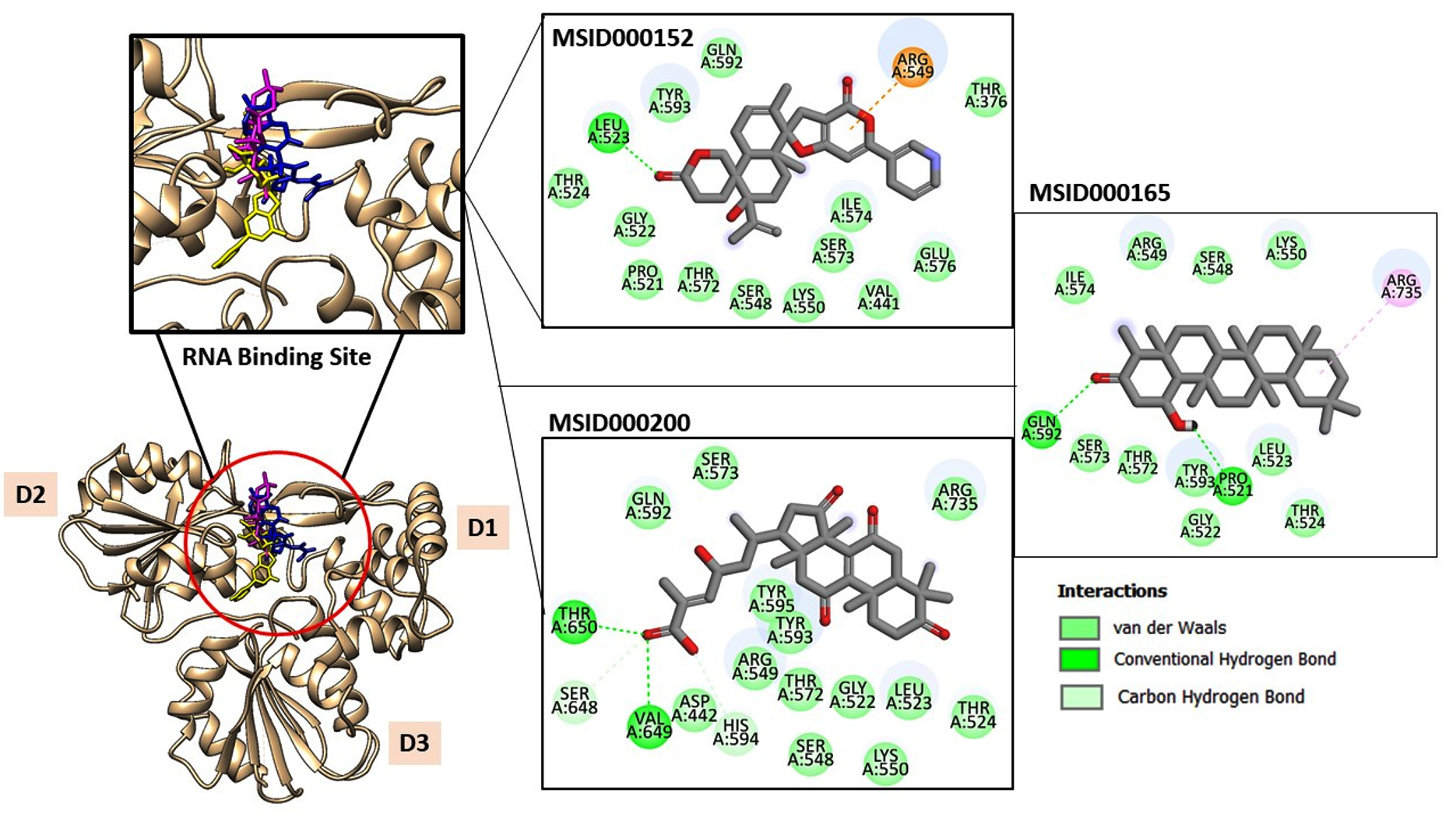
Figure 2. The docked complexes obtained after the virtual screening process. The intermolecular docked pose of NS3-like helicase enzyme with high-affinity compounds is given. The enzyme is shown by a tan cartoon with three domains labeled. The RNA binding site where all the docked compounds preferred to bind is zoom-in. The MSID000152, MSID000165, and MSID000200 are shown by yellow, blue, and magenta stick. Further, the atomic level interactions are also provided
Statistical analyses based on simulation trajectories
The dynamics of each complex were investigated within 100 ns to decipher the physical movements of the receptor enzyme in the presence of the screened inhibitors.53 All analyses were performed based on the carbon atoms of the receptor. The first analysis was the radius of gyration (Rg), which determines whether the receptor enzyme structure is compact or relaxed within the specified simulation time.54 A higher Rg value indicates a relaxed protein structure, whereas a lower Rg value indicates a compact protein structure. In the presence of these compounds, a higher Rg may lead to detachment of the compounds from the binding site and can thus be confirmed as a low-affinity binder. All the lead complexes, including the control, had lower Rg values, although some small local-level deviations were observed. The mean Rg values of MSID000152, MSID000165, MSID000200, and the control were 22.87, 24.01, 25.62, and 26.75, respectively (Figure 3A). The control complex exhibited major deviations that were attributed to the flexible loops of the enzyme. The control molecule was forced to behave more flexibly than the shortlisted leads. The lead molecules exhibited a more stable intermolecular conformation with the enzyme. The next analysis performed was the RMSD (Figure 3B).55 Similar trends were observed, with the control system being the most unstable and regular structural changes owing to the flexible loops. Among the lead systems, MSID000152 was the most stable, with a mean RMSD value of 0.95 Å. The lead complex mean RMSD ranged 1.5-2.8 Å, which signifies the high affinity of the inhibitors for the receptor helicase enzyme. Local changes in the enzyme were observed because of the same flexible loop regions reported for the control; however, they were more conformationally stable in the presence of the selected inhibitory molecules. Similarly, potent inhibitors (cyanidin 3-glucoside and dithymoquinone) of the dengue virus exhibited stable interactions with the NS2B-NS3 protease, as indicated by root mean square deviation RMSD values that ranged 2.5-3.5 Å.52 When comparing our results with their research, it is notable that compounds explored in the current study have comparable binding affinities and stability profiles. The above findings were confirmed through the root mean square fluctuation (RMSF), which indicates amino acid fluctuations versus time (Figure 3C).56 The control N-terminus showed high fluctuations compared to the lead systems. The N-terminus was the primary factor contributing to the high structural deviations in the control system. In in-lead systems, this region is stable in the presence of inhibitory compounds. The mean RMSF values for MSID000152, MSID000165, MSID000200, and the control were 0.9 6, 1.12, 1.43, and 4.10 Å, respectively. SASA analysis was performed to calculate the biomolecule surface accessible to water molecules (Figure 3D).56 Due to its relaxed nature, the control system is more exposed to interactions with water molecules than with inhibitor complexes. MSID000152 exhibited the lowest SASA scores among the lead systems.
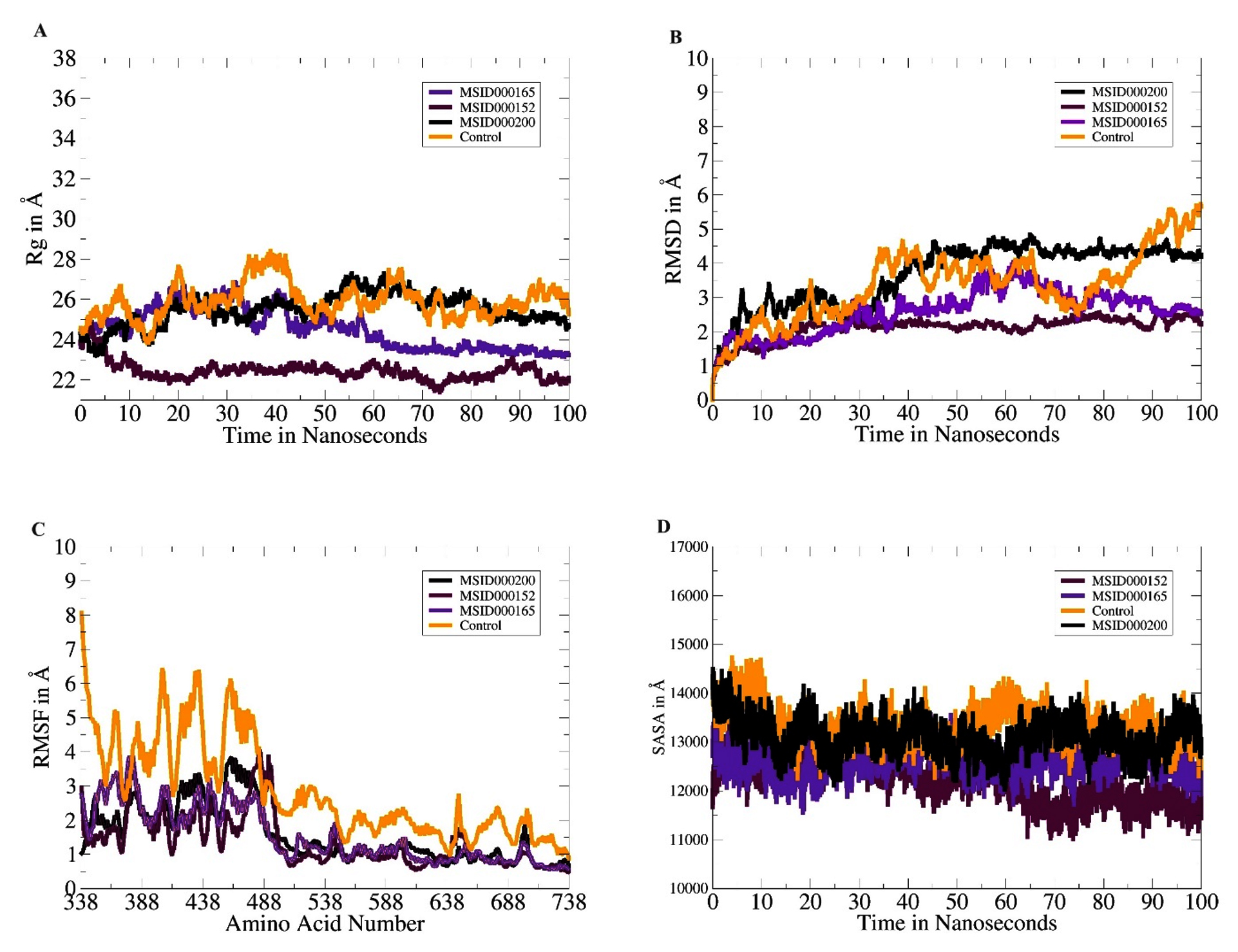
Figure 3. Molecular dynamics simulation analyses. (A). Rg, (B). RMSD, (C). RMSF and (D). SASA
Calculation of binding-free energies
Docking calculations provide a static intermolecular snapshot of the docked complex, which is not considered reliable.57 Therefore, cross-validation of the predicted binding energies is required. The MMGBSA and MMPBSA methods are popular because they are based on a series of frames collected from simulated trajectories. These methods are more accurate than docking and are modest in terms of computational power. The results obtained in the present study using the MMGBSA and MMPBSA techniques support the strong intermolecular interactions between the docked compounds and the NS3-like helicase enzyme. The order of stability of the complexes was MSID000152 > MSID000165 > MSID000200 > control. In MMGBSA, the net binding energies of MSID000152, MSID000165, MSID000200, and the control were -35.55, -33.7, -31.82, and -30.29 kcal/mol, respectively. Similarly, the MMPBSA results revealed the net binding energies of the MSID000152, MSID000165, MSID000200, and control as -35.43, -34.7, -32.34, and -31.17 kcal/mol, respectively. In each complex analyzed using MMGBSA and MMPBSA, the van der Waals interactions were identified as the dominant force in maintaining the docking of the compounds at the RNA-binding site of the enzyme. The net van der Waals interaction contribution in both MMGBSA and MMPBSA for MSID000152, MSID000165, MSID000200, and control was -36.01, -34.02, -32.08, and -30.51 kcal/mol, respectively. The contribution of columbic interaction energies was observed to be favorable in complex formations, such as -10.10 kcal/mol for MSID000152, -9.31 kcal/mol for MSID000165, -8.21 kcal/mol for MSID000200, and -8.60 kcal/mol for the control. In contrast, the solvation free energy was noted to have a non favourable contribution to complex formation. These findings suggest that the selected lead molecules showed strong binding affinities for the enzyme in both static and dynamic environments. The full statistical values obtained using MMGBSA and MMPBSA are listed in Table 2.
Table (2):
Binding interactions energies of docked complexes based on 1000 frames of simulation trajectories
| Parameter | Control | MSID000152 | MSID000165 | MSID000200 |
|---|---|---|---|---|
| MMGBSA | ||||
| Van der Waals Energy (kcal/mol) | -30.51 | -36.01 | -34.02 | -32.08 |
| Columbic Energy (kcal/mol) | -8.60 | -10.10 | -9.31 | -8.21 |
| Total Gas Phase Energy (kcal/mol) | -39.11 | -46.11 | -43.33 | -40.29 |
| Total Solvation Energy (kcal/mol) | 8.82 | 10.56 | 9.63 | 8.47 |
| Net Energy (kcal/mol) | -30.29 | -35.55 | -33.7 | -31.82 |
| MMPBSA | ||||
| Van der Waals Energy (kcal/mol) | -30.51 | -36.01 | -34.02 | -32.08 |
| Columbic Energy (kcal/mol) | -8.60 | -10.10 | -9.31 | -8.21 |
| Total Gas Phase Energy (kcal/mol) | -39.11 | -46.11 | -43.33 | -40.29 |
| Total Solvation Energy (kcal/mol) | 7.94 | 10.68 | 8.58 | 7.95 |
| Net Energy (kcal/mol) | -31.17 | -35.43 | -34.7 | -32.34 |
The values are expressed in kcal/mol
Binding entropy estimation
The presence of random energies in the selected complexes may have displaced the docked ligands. Therefore, the estimation of the entropy energy is crucial. Because the entropy energy is time-consuming, only a limited number of frames were used during the calculation. The net binding entropy energies of the docked complexes are shown in Figure 4. These results were consistent with the MMGBSA and MMPBSA results. MSID000152 was the most stable complex with the lowest random binding entropy energy. The net binding entropy of this complex was 7.5 kcal/mol. The net binding entropy energies of MSID000165 and MSID000200 were 11.2 and 14.01, respectively. All three complexes reported the lowest binding entropy energies, illustrating that each complex possessed less entropy and, therefore, less freed energy.
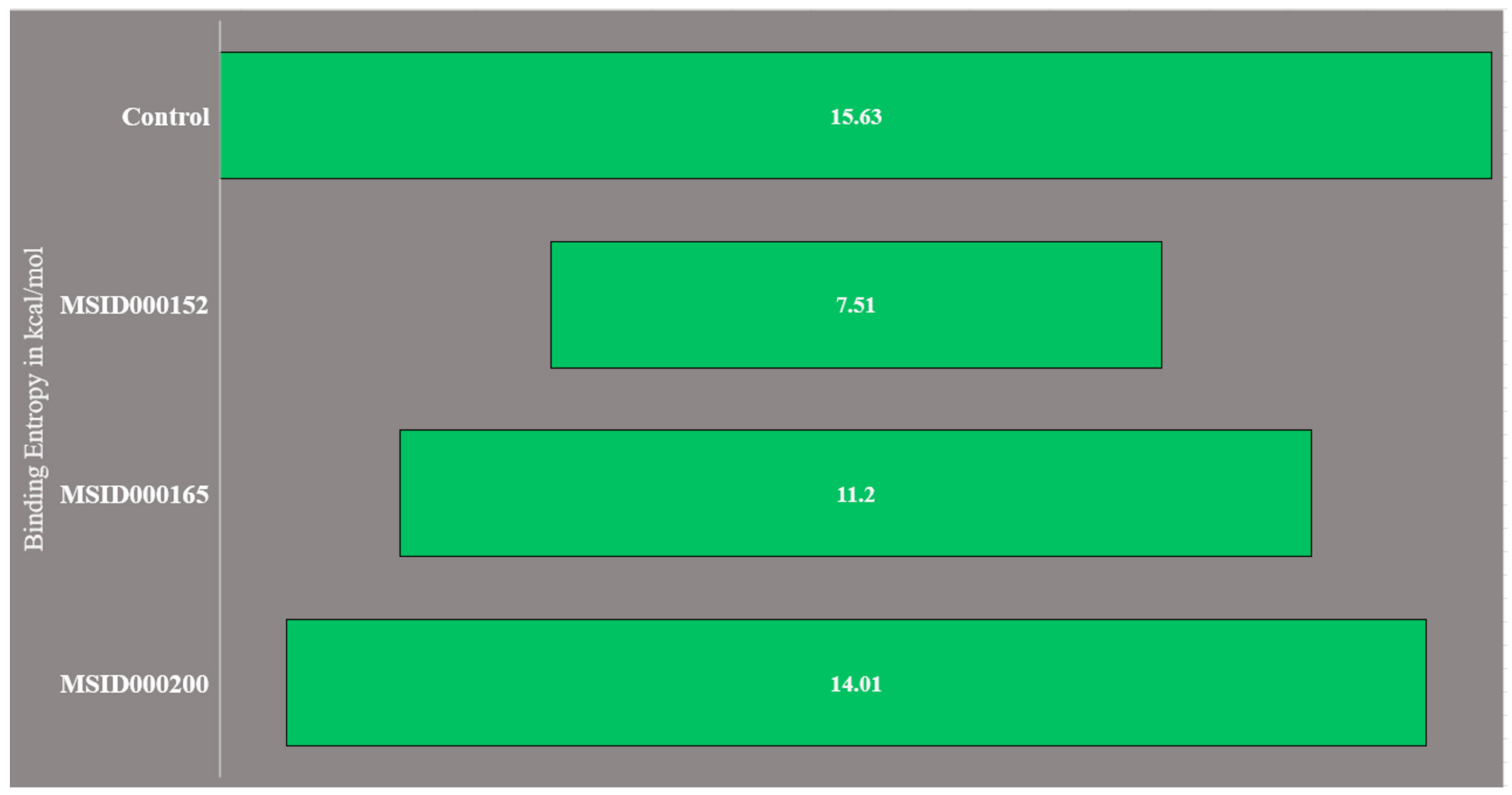
Figure 4. Binding entropy results obtained through AMBER normal mode analysis. The values are presented in kcal/mol
WaterSwap analysis
To support the experimental findings of this study thus far and revise the complex intermolecular stability and rich interaction pattern reported above, binding free energies were confirmed using the WaterSwap absolute binding free energy method.58 WaterSwap is a novel and sophisticated approach for swapping water clusters (present in the vicinity of the active pocket of the enzyme) with docked ligands. This allowed the computation of water molecules and their comparison with the ligand-binding energy. Water molecules also play a vital role in bridging ligands and enzyme-interacting residues. WaterSwap binding-energy analysis predicts the absolute binding energy using Bennetts, FEP, and TI algorithms. The WaterSwap results are shown in Figure 5. All the three complexes exhibited less deviation (<1 kcal/mol) from the values obtained using the algorithms. The MSID000152 complex exhibited the most stable energies (TI: -41.64 kcal/mol, FEP: -41.28 kcal/mol, and Bennetts: -41.25 kcal/mol).
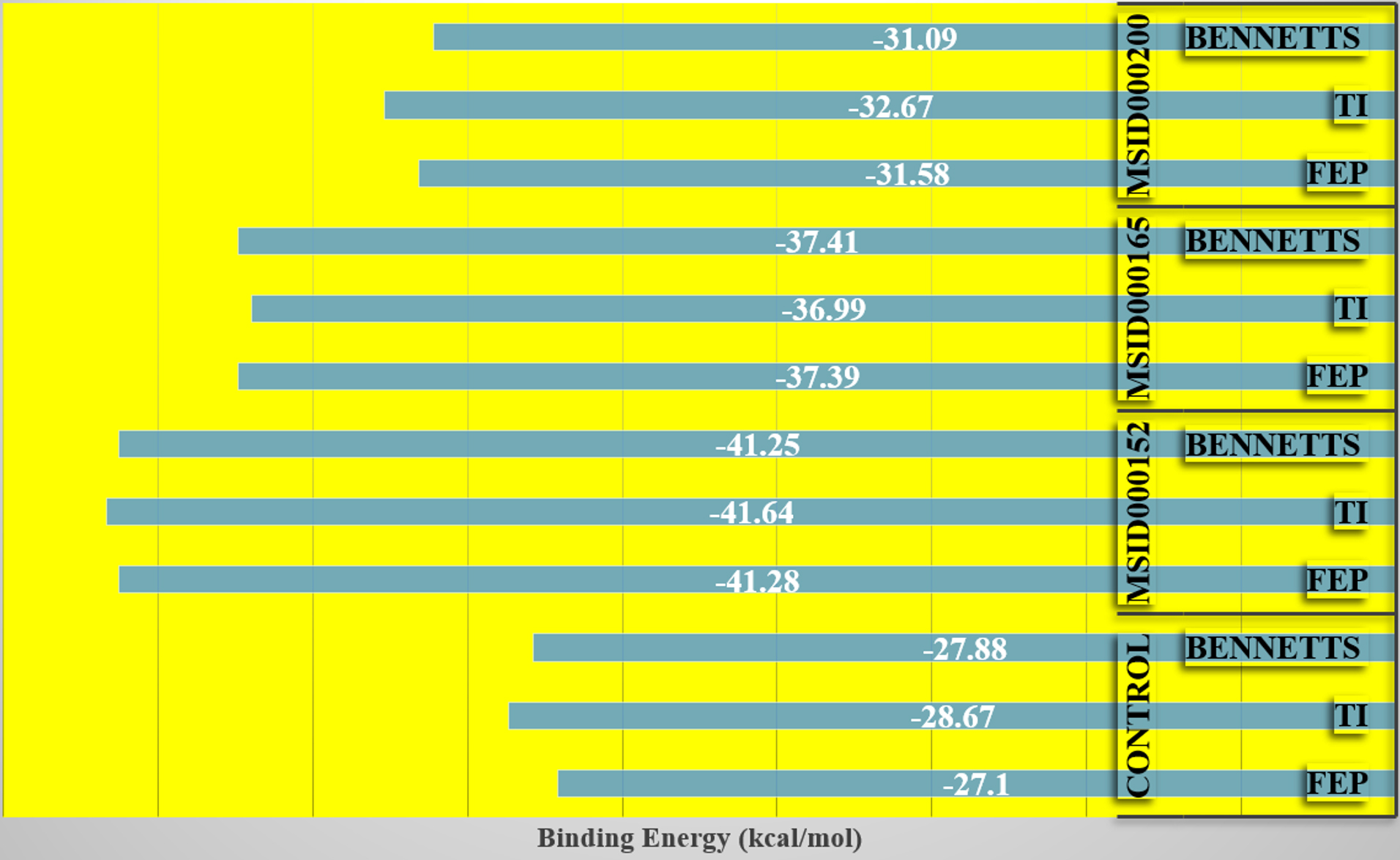
Figure 5. WaterSwap energies produced by different algorithm applied in WaterSwap analysis. Each value is plotted in kcal/mol.
Evaluation of drug-likeness of compounds
Drug-likeness is vital because drugs with therapeutic effects often fulfill different drug-like rules and have higher oral absorption than non-drug-like compounds.42 The SwissADME drug-likeness of the compounds was evaluated, and the results are shown in Table 3. Five prominent rules were used to investigate compound drug likeness: Lipinski’s rule of five, and the Ghose, Veber, Egan, and Muegge rules. The control was disclosed to adhere to these rules, and thus, may be a good candidate for branding. Among the selected compounds, MSID000152 adhered to all rules except for the Ghose rule, for which it violated two parameters: molecular weight and molar refractivity. Similarly, MSID000165 violates the Veber rule and the remaining rules. In addition, MSID000200 violated the Ghose rule. Overall, the control and all the selected leads showed good bioavailability scores.
Table (3):
Evaluation of control and selected inhibitory compounds in drug-likeness by SwissADME
Drug Rule |
Control |
MSID000152 |
MSID000165 |
MSID000200 |
|---|---|---|---|---|
Lipinski Rule of Five |
Yes; 0 violation |
Yes; 0 violation |
Yes; 1 violation: MLOGP>4.15 |
Yes; 0 violation |
Ghose |
Yes |
No; 2 violations: MW>480, MR>130 |
No; 3 violations: WLOGP>5.6, MR>130, #atoms>70 |
No; 3 violations: MW>480, MR>130, #atoms>70 |
Veber |
Yes; 0 violation |
Yes |
Yes |
Yes |
Egan |
Yes; 0 violation |
Yes |
No; 1 violation: WLOGP>5.88 |
Yes |
Muegge |
Yes; 0 violation |
Yes |
No; 1 violation: XLOGP3>5 |
Yes |
Bioavailability Score |
0.56 |
0.55 |
0.55 |
0.56 |
Evaluation of pharmacokinetic properties
The pharmacokinetic properties of selected compounds were determined. This analysis is vital for elucidating the ADMET of a compound. The pharmacokinetics of MSID000152, MSID000165, and MSID000200 are shown in Figure 6, Supplementary Table 1, and Supplementary Table 2, respectively. The pharmacokinetic properties of high-affinity binders are presented in the main text. MSID000152 exhibited the highest intestinal absorption rate (97%). The compound exhibited a weak ability to cross the blood-brain barrier, and thus did not cause any adverse central nervous system side effects. The compound also showed no AMES or low minnow toxicity. The oral absorption of the compound was high; thus, a good magnitude of the drug reached the target side for therapeutic effect.59 This compound has no skin sensitization ability and is predicted to be easily cleared from the body. The remaining pharmacokinetic properties are shown in Figure 6. Computational approaches have several limitations, including dependence on database accuracy and inherent presumptions of in silico models. Molecular docking and ADME predictions do not fully capture in vivo complexities.60 Therefore, further experimental studies are required to validate the therapeutic potential of these inhibitors.
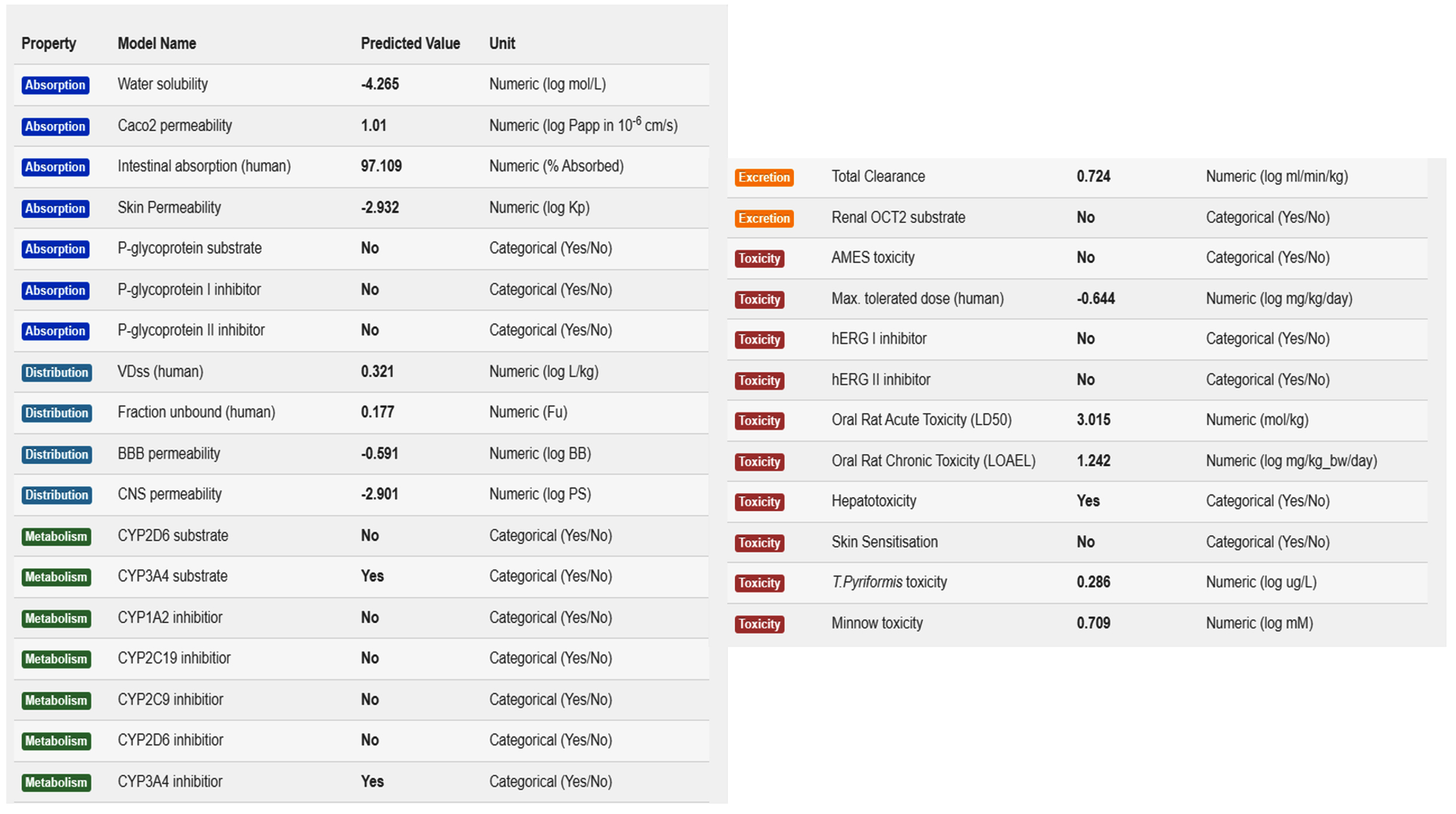
Figure 6. Pharmacokinetic properties of compound MSID000152.
This study used diverse biophysics-, machine learning-, and artificial intelligence-supported servers and software to identify five novel natural inhibitors (MSID000152, MSID000165, MSID000200, AfroDb.28, and AfroDb.207) of NS3-like helicase enzymes from the Alongshan virus. The inhibitors showed a significant affinity for the RNA-binding site of the enzyme and revealed a strong and rich interaction network. Furthermore, the compounds showed stable dynamics, dominant van der Waals, and electrostatic interactions. The entropy energy of each complex is low; thus, it has a low-disorder energy. These compounds are also good candidates because of their drug-likeness and pharmacokinetic properties. Therefore, these compounds can be used in different experimental biological tests to validate in silico predictions. This investigation is useful for understanding the binding affinities of NS3-like helicase inhibitors in the search for novel and better antiviral drugs against the Alongshan virus. Furthermore, the exploration of natural compounds and their interactions with viral enzymes has potential in the development of more effective antiviral therapies.
Additional file: Additional Table S1-S2.
ACKNOWLEDGMENTS
None.
FUNDING
None.
DATA AVAILABILITY
All datasets generated or analyzed during this study are included in the manuscript.
ETHICS STATEMENT
Not applicable.
- Zhao Y, Sui L, Pan M, et al. The segmented flavivirus Alongshan virus reduces mitochondrial mass by degrading STAT2 to suppress the innate immune response. bioRxiv. 2024;e01301-24.
Crossref - Kholodilov IS, Belova OA, Morozkin ES, et al. Geographical and tick-dependent distribution of flavi-like Alongshan and Yanggou tick viruses in Russia. Viruses. 2021;13(3):458.
Crossref - Gomer A, Lang A, Janshoff S, Steinmann J, Steinmann E. Epidemiology and global spread of emerging tick-borne Alongshan virus. Emerg Microbes Infect. 2024;13(1):2404271.
Crossref - Zhang X, Wang N, Wang Z, Liu Q. The discovery of segmented flaviviruses: implications for viral emergence. Curr Opin Virol. 2020;40:11-18.
Crossref - Gao X, Zhu K, Wojdyla JA, et al. Crystal structure of the NS3-like helicase from Alongshan virus. IUCrJ. 2020;7(3):375-382.
Crossref - Gladysheva AA, Gladysheva AV, Ternovoi VA, Loktev VB. Structural Motifs and Spatial Structures of Helicase (NS3) and RNA-dependent RNA-polymerase (NS5) of a Flavi-like Kindia tick virus (unclassified Flaviviridae). Vopr Virusol. 2023;68(1):7-17.
Crossref - Wang Z-D, Wang W, Wang N-N, et al. Prevalence of the emerging novel Alongshan virus infection in sheep and cattle in Inner Mongolia, northeastern China. Parasit Vectors. 2019:12:1-7.
Crossref - Beltz LA. Zika and Other Neglected and Emerging Flaviviruses-E-Book: The Continuing Threat to Human Health. Elsevier Health Sciences: 2021.
Crossref - Bassani D, Moro S. Past, present, and future perspectives on computer-aided drug design methodologies. Molecules. 2023;28(9):3906.
Crossref - Yu W, MacKerell AD. Computer-aided drug design methods. Methods Mol Biol. 2017;1520:85-106.
Crossref - Vemula D, Jayasurya P, Sushmitha V, Kumar YN, Bhandari V. CADD, AI and ML in drug discovery: A comprehensive review. Eur J Pharm Sci. 2023;181:106324.
Crossref - Durrant JD, McCammon JA. Computer-aided drug-discovery techniques that account for receptor flexibility. Curr Opin Pharmacol. 2010:10(6):770-774.
Crossref - Talele TT, Khedkar SA, Rigby AC. Successful applications of computer aided drug discovery: moving drugs from concept to the clinic. Curr Top Med Chem. 2010;10(1):127-141.
Crossref - Durrant JD, McCammon JA. Molecular dynamics simulations and drug discovery. BMC Biol. 2011;9:71.
Crossref - Massova I, Kollman PA. Combined molecular mechanical and continuum solvent approach (MM-PBSA/GBSA) to predict ligand binding. Perspect Drug Discov Des. 2000;18(1):113-135.
Crossref - Alamri MA, Tariq MH, Ul Qamar MT, Alabbas AB, Alqahtani SM, Ahmad SJ. Dynamics, Discovery of potential phytochemicals as inhibitors of TcdB, a major virulence factors of Clostridioides difficile. J Biomol Struct Dyn. 2023;41(22):12768-12776.
Crossref - Cheng F, Li W, Zhou Y, et al. admetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties. J Chem Inf Model : 2012;52(11):3099-3105.
Crossref - Berman HM, Westbrook J, Feng Z, e al. The protein data bank. Nucleic Acids Res. 2000;28(1):235-242.
Crossref - Ahmad S, Waheed Y, Abro A, Abbasi SW, Ismail S. Molecular screening of glycyrrhizin-based inhibitors against ACE2 host receptor of SARS-CoV-2. J Mol Model. 2021;27(7):206.
Crossref - Ntie-Kang F, Zofou D, Babiaka SB, et al. AfroDb: a select highly potent and diverse natural product library from African medicinal plants. PLoS One. 2013;8(10):e78085.
Crossref - Vivek-Ananth RP, Sahoo AK, Kumaravel K, Mohanraj K, Samal A. MeFSAT: a curated natural product database specific to secondary metabolites of medicinal fungi. RSC Adv. 2021;11(5):2596-2607.
Crossref - Valdes-Jimenez, A, Pena-Varas, C, Borrego-Munoz P, et al. PSC-db: a structured and searchable 3D-database for plant secondary compounds. Molecules. 2021;26(4):1124.
Crossref - Dallakyan S, Olson AJ. protocols, Small-molecule library screening by docking with PyRx. Methods Mol Biol. 2015:243-250.
Crossref - Ounthaisong U, Tangyuenyongwatana P. Cross-docking study of flavonoids against tyrosinase enzymes using PyRx 0.8 virtual screening tool. Thai Journal of Pharmaceutical Sciences. 2017;41(Suppl Issue):189-192.
- Bommu UD, Konidala KK, Pamanji R, Yeguvapalli S. Computational screening, ensemble docking and pharmacophore analysis of potential gefitinib analogues against epidermal growth factor receptor. J Recept Signal Transduct Res. 2018;38(1):48-60.
Crossref - Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera-a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605-1612.
Crossref - Sharma S, Sharma A, Gupta U. Molecular Docking studies on the Anti-fungal activity of Allium sativum (Garlic) against Mucormycosis (black fungus) by BIOVIA discovery studio visualizer 21.1.0.0. Ann Antivir Antiretrovir. 2021;12(5):028-32.
Crossref - Naqvi AAT, Mohammad T, Hasan GM, Hassan MI. Advancements in docking and molecular dynamics simulations towards ligand-receptor interactions and structure-function relationships. Curr Top Med Chem. 2018;18(20):1755-1768.
Crossref - Case DA, Duke RE, Walker RC, et al. AMBER 22 reference manual. 2022.
Crossref - Tian C, Kasavajhala K, Belfon KAA, et al. ff19SB: amino-acid-specific protein backbone parameters trained against quantum mechanics energy surfaces in solution. J Chem Theory Comput. 2019;16(1):528-552.
Crossref - Ismail S, Ahmad S, Azam SS. Vaccinomics to design a novel single chimeric subunit vaccine for broad-spectrum immunological applications targeting nosocomial Enterobacteriaceae pathogens. Eur J Pharm Sci. 2020;146:105258.
Crossref - Xiong Y, Shabane PS, Onufriev AV. Melting points of OPC and OPC3 water models. ACS Omega. 2020;5(39):25087-25094.
Crossref - Abad SFG. Molecular Dynamics and Nonlinear Dynamics Studies of Chemical Systems. West Virginia University: 2018.
- Abbasi S, Raza S, Azam SS, Liedl KR, Fuchs JE. Interaction mechanisms of a melatonergic inhibitor in the melatonin synthesis pathway. J Mol Liq. 2016;221:507-517.
Crossref - Roe DR, Cheatham III TE. PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J Chem Theory Comput. 2013;9(7):3084-3095.
Crossref - Yan D, Yang Y, Shen H, Liu Z, Yao K, Liu Q. 3D-QSAR and Molecular Dynamics Study of Isoxazole Derivatives to Identify the Structural Requirements for Farnesoid X Receptor (FXR) Agonists. Molecules. 2024;29(6):1210.
Crossref - Miller III BR, McGee Jr TD, Swails JM, Homeyer N, Gohlke H, Roitberg AE. MMPBSA.py: an efficient program for end-state free energy calculations. J Chem Theory Comput. 2012;8(9):3314-3321.
Crossref - Genheden S, Ryde U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin Drug Discov. 2015;10(5):449-461.
Crossref - Ding Y, Tang J, Guo F. Identification of protein-ligand binding sites by sequence information and ensemble classifier. J Chem Inf Model. 2017;57(12):3149-3161.
Crossref - Genheden S, Kuhn, O, Mikulskis P, Hoffmann D, Ryde U. The normal-mode entropy in the MM/GBSA method: effect of system truncation, buffer region, and dielectric constant. J Chem Inf Model. 2012;52(8):2079-2088.
Crossref - Woods CJ, Malaisree M, Michel J, Long B, McIntosh-Smith S, Mulholland AJ. Rapid decomposition and visualisation of protein-ligand binding free energies by residue and by water. Faraday Discuss. 2014;169:477-499.
Crossref - Bergstrom CAS, Larsson P. Computational prediction of drug solubility in water-based systems: Qualitative and quantitative approaches used in the current drug discovery and development setting. Int J Pharm. 2018;540(1-2):185-193.
Crossref - Lipinski CA. Lead-and drug-like compounds: the rule-of-five revolution. Drug Discov Today Technol. 2004;1(4):337-341.
Crossref - Sardar H. Drug like potential of Daidzein using SwissADME prediction: In silico Approaches. Phytonutrients. 2023:02-08.
- Egan WJ, Merz KM, Baldwin JJ. Prediction of drug absorption using multivariate statistics. J Med Chem. 2000;43(21):3867-3877.
Crossref - Veber DF, Johnson SR, Cheng H-Y, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002;45(12):2615-2623.
Crossref - Muegge I, Heald SL, Brittelli D. Simple selection criteria for drug-like chemical matter. J Med Chem2001;44(12):1841-1846.
Crossref - Pires DEV, Blundell TL, Ascher DB. pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J Med Chem. 2015;58(9):4066-4072.
Crossref - Van De Waterbeemd H, Gifford E. ADMET in silico modelling: towards prediction paradise? Nat Rev Drug Discov. 2003;2(3):192-204.
Crossref - Singh V, Somvanshi P. Structural Modeling of the NS 3 helicase of Tick-borne encephalitis virus and their virtual screening of potent drugs using molecular docking. Interdiscip Sci. 2009;1(3):168-172.
Crossref - Ejeh S, Uzairu A, Shallangwa G, Abechi S, Ibrahim MT. In Silico Identification of Some Novel Ketoamides as Potential Pan-Genotypic HCV NS3/4A Protease Inhibitors with Drug-Likeness, Pharmacokinetic ADME Profiles, and Synthetic Accessibility Predictions. Adv J Chem A. 2022;5(3):197-207.
Crossref - Rahman MM, Biswas S, Islam KJ, et al. Medicine, Antiviral phytochemicals as potent inhibitors against NS3 protease of dengue virus. Comput Biol Med. 2021;134:104492.
Crossref - Hansson T, Oostenbrink C, van Gunsteren W. Molecular dynamics simulations. Curr Opin Struct Biol. 2002;12(2):190-196.
Crossref - Lobanov MY, Bogatyreva NS, Galzitskaya OV. Radius of gyration as an indicator of protein structure compactness. Mol Biol. 2008;42(4):623-628.
Crossref - Alamri MA, Ahmad S, Alqahtani SM, Irfan M, Alabbas AB, Ul Qamar MT. Dynamics, Screening of marine natural products for potential inhibitors targeting biotin biosynthesis pathway in Mycobacterium tuberculosis. J Biomol Struct Dyn. 2023;41(17):8535-8543.
Crossref - Ahmad S, Raza S, Uddin R, Azam SS. Binding mode analysis, dynamic simulation and binding free energy calculations of the MurF ligase from Acinetobacter baumannii. J Mol Graph Model. 2017;77:72-85.
Crossref - Wang E, Sun H, Wang J, et al. End-point binding free energy calculation with MM/PBSA and MM/GBSA: strategies and applications in drug design. Chem Rev. 2019;119(16):9478-9508.
Crossref - Woods CJ, Malaisree M, Hannongbua S, Mulholland AJ. A water-swap reaction coordinate for the calculation of absolute protein-ligand binding free energies. J Chem Phys. 2011;134(5).
Crossref - Protti IF, Rodrigues DR, Fonseca SK, Alves RJ, de Oliveira RB, Maltarollo VG. Do drug likeness rules apply to oral prodrugs? ChemMedChem. 2021;16(9):1446-1456.
Crossref - Pelkonen O, Turpeinen M, Raunio H. In vivo-in vitro-in silico pharmacokinetic modelling in drug development: current status and future directions. Clin Pharmacokinet. 2011;50:483-491.
Crossref
© The Author(s) 2025. Open Access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License which permits unrestricted use, sharing, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.