


The quantity and diversity of the microbial community in soil make it possibly the most difficult of all the natural ecosystems. It is thought to be challenging to culture up to 99% of the microorganisms in a given environment. The intricacy of microbial variety is impacted by numerous interconnected factors, including as soil structure, water content, biotic activity, pH, and fluctuations in climate. Environmental DNA isolation and purification are often the first steps in the soil metagenomic analysis process. Creating genomic DNA libraries and then using them for high-throughput sequencing or library screening are the main steps in the application of metagenomics. These genomic sequences are currently being used to advance our knowledge of the ecology and physiology of these bacteria as well as for new biotechnological and medicinal applications. To completely comprehend the intricacies involved in the operation of microbial communities and the interactions between different microorganisms within specific niches, metagenomic sequences are employed. This study focuses on the latest advancements in biotechnological approaches and procedures for identifying novel genes from uncultured microorganisms and intricate microbial habitats.
Metagenomics, Humic Compounds, Novel Compounds, Metagenomic Libraries, Phylogenetic Markers
Microbial populations inhabiting the environment like soil, water, and air may not be accessible as they are unculturable in the laboratory by conventional methods used in the laboratory.1 On Earth, there are thought to be 1030 microbiological cells, with prokaryotes accounting for the majority of individual organisms, which comprise 106 to 108 distinct genospecies.2,3 Only 0.1-1% of all microorganisms found in nature can be grown in a typical laboratory setting.4 Therefore, using conventional culturable techniques, it is hard for researchers to study more than 99% of the microorganisms that exist-microbes that can occasionally have peculiar but potentially extremely helpful characteristics like breaking down garbage or synthesising new substances like antibiotics or pharmaceuticals. Without the requirement for culture, researchers can examine wild microbial populations through the analysis of directly extracted nucleic acids from ambient samples. Due to the development of nucleic acid-based technology, the taxonomy based on routine parameters such as morphological, physiological, and biochemical was no longer useful or authentic.5 The metagenome is made up of all the genetic material found in environmental samples that is made up of the genomes of numerous different organisms. DNA sequences have been accumulated as a result of metagenomics, and these sequences are being used for innovative biotechnological applications. The field of metagenomics is a technique that enables the examination of the great diversity of individual genes and their products as well as the analysis of complete operons encoding metabolic, biosynthetic, or biodegradative processes.6,7 It involves the extraction, cloning, functional screening, and direct random shotgun sequencing of the entire genetic complement of habitat.8-10 The uncultured microorganism’s genomes comprise vast quantities of data, and one of the most advanced methods to discover and investigate this potential is metagenomics. Pharmaceuticals, agrochemicals, and fine chemicals are all produced using metagenomics technology, since the advantages of chiral synthesis catalysed by enzymes are becoming more widely acknowledged. Finding relevant natural and synthetic compounds for medication development is now more likely because to the recovery of metabolic pathway gene clusters involved in the manufacture of antibiotics and bioactive chemicals.11,12 One of the hardest samples to work with when developing appropriate extraction and purification techniques is soil. Many substances found in the complex soil matrix interfere with hybridization and detection protocols, the probable reason is polymerases and restriction enzymes inhibition.13 Of particular concern are the highly abundant natural degradation-prone humic and fulvic acids found in high-organic soil. These fractions exhibit a wide range of solubilities and charge properties because they are such intricate combinations of related chemicals. It is difficult to find extraction and separation techniques that can remove all humic chemicals from soil DNA samples, if not impossible. A culture-dependent method has been employed more frequently in recent years to supplement two or more culture-independent methods in order to lessen biases brought on by using only one method. For instance, two culture-independent methods were used to study the bacterial communities of the nostril and posterior wall of the oropharynx. Microarray and 16S rRNA gene clone library analyses revealed similar phylogenetic distribution patterns, indicating good concordance between these profiling approaches.14 Numerous techniques have been developed to extract high-quality DNA from a range of environmental materials, such as surface water from rivers,15 polluted subterranean sediments, groundwater,16,17 marine picoplankton,18 soil,19-26 hot springs and mud holes in solfataric fields,27 glacier ice,28 buffalo rumens,29 and Antarctic desert soil.30 The most current advancements in biotechnological methods and procedures for locating new genes in unexplored microbial niches and reservoirs of microorganisms are covered in this review.
Metagenomics: a tool for novel compounds
Numerous possible uses for metagenomics exist in the field of biotechnology (Figure 1). Using a shotgun sequencing method, researchers have sampled the genetic content of these diverse habitats using soil,6 acid mine drainage,31 and varied environments.32 Using advanced techniques such as gene arrays, proteomics-based analysis, and microscopy, metagenomics unlocks information about the unexplored microbial community that is currently not accessible to unculturability of all microorganisms. Basic and applied approaches for metagenomics have explored microbial diversity, evolutionary relationship, genetic, population structure, functional activity, relationships with the environment, the discovery of antibiotics, and industrially important enzymes.33
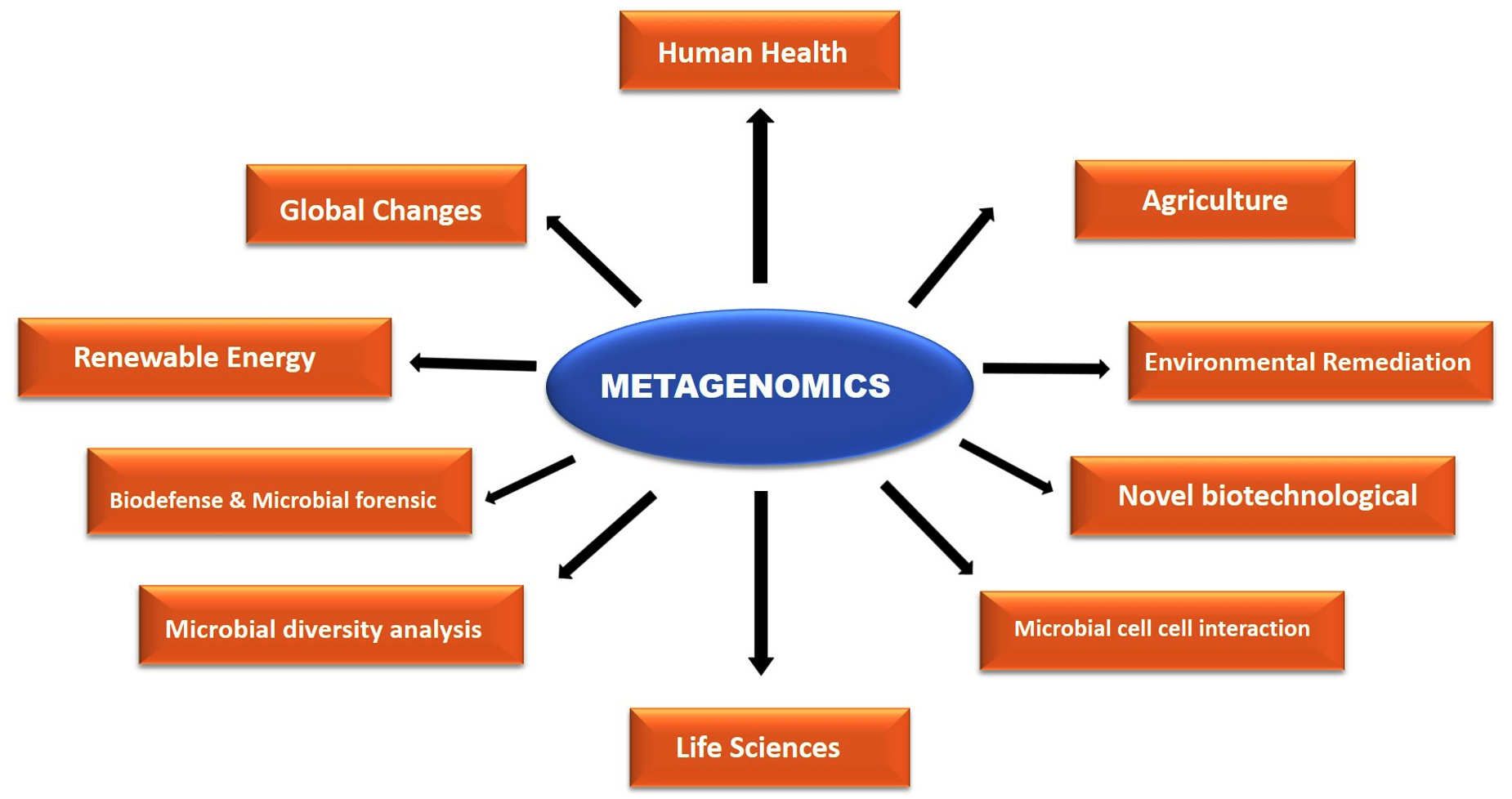
Figure 1. Applied applications of metagenomics
Through metagenomics, a number of new genes and gene products are found, such as the first bacteriorhodopsin of bacterial origin, new members of protein families that are already known, like DNA Polymerase, RecA, and Na+(Li+)/H+ antiporters, as well as determinants of antibiotic resistance and small molecules with antimicrobial activity.34-36
Approaches to metagenomics
The metagenomics involves the quality DNA isolation from environmental samples, cloned into an appropriate vector, introduced into the host bacterium, and the resulting transformants are screened (Figure 2). Clones can be randomly sequenced, screened for phylogenetic markers like 16S rRNA and rec A, or screened for the expression of certain features like enzyme activity or antibiotic synthesis using multiplex PCR or hybridization.37 While there are advantages and disadvantages to each metagenomic technique, taken as a whole, they have improved our knowledge of non-culturable microbes and shed light on previously unidentified prokaryotic groupings.
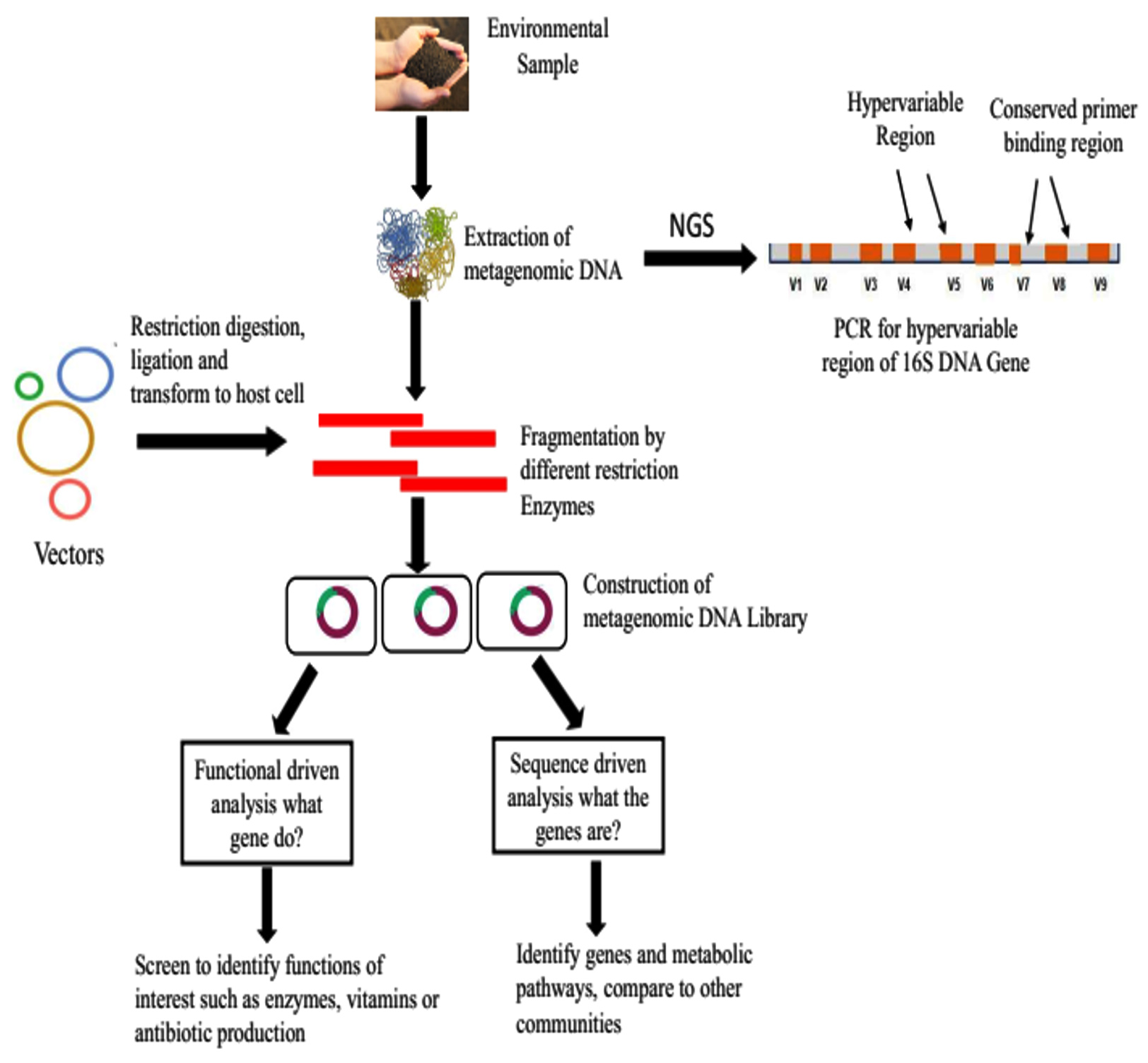
Figure 2. Metagenomic approach
Analysis on the basis of sequence
Sequence-based analysis can be used to perform complete sequencing of clones with phylogenetic anchors indicating the taxonomic group most likely to be the source of the DNA fragment. Based on conserved DNA sequences, sequence-based screening is not affected by the expression of cloned genes in a heterologous host. The creation of metagenomic libraries and shotgun sequencing have made a substantial amount of data available, including millions of novel genes, phylogenetic relationships, and predicted metabolic pathways of non-culturable bacteria.38 Random clones can be sequenced using the phylogenetic marker-driven technique, yielding striking findings. Over 210 distinct metagenomes had been sequenced from a wide range of habitats, soil, human guts, faeces, and worldwide oceans.21 Large-scale microbial niche study is now possible because of next-generation sequencing techniques, which have prompted the creation of novel applications including metabolomics, meta-transcriptomics, and comparative community metagenomics.39 As an authentic photo-receptor, bacteriorhodopsin-like genes have also been investigated by the metagenomic investigation. These genes are found abundantly among the ocean’s proteobacteria, not just in archaea.40,41 The process of reconstructing the genomes of organisms that have never been cultivated in an acid mine drainage community is an example of how large-scale sequencing efforts can enhance our knowledge of uneducated populations.42 A metagenomic study of river Ganga explored the bacterial diversity and found Erythrobacter litoralis, Novosphingobium aromaticivorans, and Sphingopyxis alaskensis are the most abundant bacteria. Through these investigations, the relationships between phylogeny and function, the prevalence of particular gene types, and the reconstruction of non-culturable species’ genomes have all been developed.
Function driven approach
Extraction of DNA, library construction, and functional screening
Sample handling and cell lysis
The preparation of samples before DNA extraction is crucial for researching the microbial ecology of natural populations. It is crucial to extract DNA as soon as possible from the fresh samples. Prior to extractions, samples can also be kept for hours or even days at 4°C. To lessen the impact of nuggets, proper mixing is required, and extraction should be carried out on grams of material.43
The isolation of ambient DNA typically serves as the starting point for metagenome investigation. Metagenomic DNA isolation from soil and sediment samples can be classified into two primary categories: direct and indirect extraction approaches. Direct DNA isolation is predicated on cell lysis inside the sample matrix and subsequent DNA separation from the cell debris.44 The indirect method entails removing the cells from the soil matrix, lysing the cells, and then extracting the DNA.45
Cell lysis is the most important stage in the extraction of soil metagenomic DNA. Its goal is to rupture the cell wall and membrane of microorganisms in order to liberate the DNA.46 The main problem encountered for soil or sediment samples that while chemical or enzymatic lysis the penetration is poor and it also depend on cell types. Mechanical disruption is more effective than chemical or enzymatic lysis as it provides more uniform disruption of cells, soil and sediment samples that allows excellent lysis buffer penetration. As a result, compared to chemical lysis, mechanical treatment is less selective and more effective.47 Thermal shocks, or repeated freezing and thawing, bead-mill homogenization, bead-beating, microwave heating, and ultrasonication are examples of mechanical disruption techniques. It is possible to alter the number of freeze-thaw cycles, as well as the incubation period and temperature.48,49 Compared to other mechanical treatments such as bead beating, ultrasonication, and microwave heating, thermal shock is somewhat less violent.50 The bacterial cells attached to the soil aggregates are released by the effectiveness of the ultrasonication therapy.51 Cell lysis can be achieved very successfully by ultrasonication, microwave heating, and thermal shocks.52 It was suggested that in order to completely lyse the Streptomyces spores, these three treatments have to be administered. DNA is sheared as a result of mechanical treatments such bead pounding and sonication. Longer beating periods, faster speeds, and smaller volumes of extraction buffer all resulted in higher DNA yields. But there was always a corresponding rise in DNA shearing with higher DNA output.53 There are 63-81% lysis reported using different techniques, up to 90% lysis efficiency.54-56 By using viable and direct cell counts, showed lysis efficiency ranging from 25 to 66% following grinding and 74% following bead-beating.57 The amount of clay in the soil had a negative correlation with the lysis efficiency.58
Many protocols employ a combination of physical and chemical methods. The most common detergent treatment consists of 1% SDS and salt concentrations of 1 M or higher, frequently combined with heating and shaking.50 Since DNA tends to sink into soil particles, resulting in poorer yields, a hot-SDS lysis method was initially introduced by soil extraction.59,60 Although SDS can hinder PCR if it is not eliminated in later steps, adding detergents and salts can help to mitigate this issue.61 SDS is the most frequently used detergent for cell lysis.62 Some Gram-positive bacteria, however, might not be lysed by the SDS-based lysis method. For the majority of Gram-positive bacteria, the DNA yield was found to be two to six times greater using a process that involved grinding, freezing, and thawing, followed by SDS-based lysis.56 Combining chemical and mechanical lysis method greater the yield.63 They discovered that, compared to bead-beating or lysis at 70°C alone, homogenization in a bead-beater for one minute and one hour of incubation at
70°C in high salt-SDS buffer produced twice as much DNA.
Metagenomic DNA extraction and purification
Soil homogenates can be centrifuged to remove soil debris, but often the extractions proceed directly with the homogenate. Many of the methods facilitate breakage and subsequent deproteination by adding CTAB and increasing the salt concentration.47,56,58 Proteins have been salted using saturated salt solution NaCl,64 ammonium acetate,65 and sodium acetate.45 Nucleic acids were recovered in the supernatant after low-speed centrifugation precipitated proteins. NaCl was shown to deproteinize protein.66 The extraction buffer’s pH has a significant impact on the recovery of soil metagenomic DNA. Twenty distinct soil samples were used to optimize the pH of the DNA-extraction buffer.57 At pH 9-10, the maximum yield of DNA was produced. Nonetheless, at pH 10 compared to pH 9, more humic materials were released. Polyethylene glycol (PEG) has been shown to improve soil metagenomic DNA precipitation.67,68 Humic acid removal may also be aided by the extraction buffer that contains 1.2% CTAB and 4.5% NaCl, which is then incubated for 20 minutes at 65oC.43 Extracted DNA with good quality and quantity can be obtained by utilizing lysis buffer and heat treatment at 65°C for genomic DNA extraction.69 The precipitation of soil metagenomic DNA with PEG 8000 or isopropanol resulted in a higher recovery of humic chemicals and a poorer recovery of DNA. However, phenol extraction aids in the removal of PEG 8000 since it interferes with PCR. It was suggested that 5% PEG be used for the precipitation of soil metagenomic DNA since it produced noticeably fewer humic acids during the precipitation process without having an impact on PCR.70 The crude DNA extracts from soils typically require additional purification because they are typically too contaminated for molecular analysis. The ambient DNA could not be effectively purified using any approach. The most common purification method for removing humic components from DNA extracts is the use of silica gel or silica membrane separation.56,71,72 DEAE-cellulose columns are frequently used for purifying tRNAs and have been employed for DNA extracts. The silica gel first binds to humic material and DNA, and some humic acids can be sequentially eluted from the matrix.73 Ion exchange chromatography and hydroxyapatite column chromatography are effective methods for removing a significant number of humic compounds. DNA may be effectively separated from humic materials using agarose gel electrophoresis.56,74 DNA is also separated according to size using polyacrylamide and dextran gel filtration columns.50,56,74-78 Sepharose 4B was found to be more successful for excellent separation when tested the effectiveness of Sephadex G-200, Sephadex G-50, and Sepharose 4B with a variety of soils.75 When extracting DNA for purification, the cesium chloride gradient-a traditional technique-seems to be quite successful.79-82 Humic materials can also be eliminated using the process of differential ethanol precipitation. The collaboration of chromatographic and spectroscopic methods with NGS makes the sequencing process less error prone and more efficient in gene expression profiling.83
Quantification of metagenomic DNA
Humic compounds found in soil metagenomic DNA sometimes cause problems when quantifying DNA. Humic compounds have a molecular mass ranging from 0.1 to >300 KDa, are dark brown in color, structurally complex, polyphenolic, and polyelectrolytic. Based on their solubility in acid or alkali, humic compounds can be classified into three main fractions: (i) humic acid, which is both alkali-soluble and acid insoluble; (ii) fulvic acid, which is both alkali and acid soluble; and (iii) humin, which is both alkali and acid insoluble.84 Contamination by humic molecules, polyphenolic ions, thiocyanates, and other organic compounds might result in absorption at 230 nm. A260/A230 for pure DNA samples should be close to 1.8. Phenol contaminates DNA samples, is frequently employed in nucleic acid purification, and can greatly alter quantification estimates. Phenol absorbs with an A260/A280 ratio of 1.2 with a peak at 270 nm. The quantities of humic chemicals, not DNA, are shown by absorption at 260 nm.75 Densitometric measurement of an agarose gel stained with ethidium bromide can be used to assess the content of DNA.85 Using the fluorescent dye picoGreen, soil metagenomic DNA may be quantified effectively. The double-stranded DNA is bound by the dye, and a fluorometer is used to measure the DNA-picoGreen complex. PicoGreen fluorescence is disrupted by sample DNA that has more than 100 ng/µl of humic acid in it. Double-stranded DNA can be quantified down to 500 pg/ml with PicoGreen. However, humic acid cannot tamper with DNA measurement at amounts less than 10 ng/µl. With this technique, crude DNA extracted straight from the soil can be measured without the need for a purification phase.4
Determination of yield and quality of DNA
Humic compounds are created during the degradation of plant, animal, and microbial biomass in soil metagenomic DNA isolation. DNA absorbs at 260 nm, while humic compounds, which have a complicated structure, absorb at 230 nm. Accordingly, the purity of the soil metagenomic DNA is often assessed using absorbance ratios at 260/230 nm (DNA/humic acid) and 260/280 nm (DNA/protein).62 Humic compounds are created during the degradation of plant, animal, and microbial biomass in soil metagenomic DNA isolation. DNA absorbs at 260 nm, while humic compounds, which have a complicated structure, absorb at 230 nm. DNA yield is calculated by measuring the absorbance at 260 nm of the elute to estimate the concentration of DNA; the absorbance should fall between 0.1 and 1.0. The presence of other elements, such as contaminating particulate particles in a filthy cuvette, is indicated by absorbance at 325 nm. For a 1 cm detection route, a 260 nm value of 1 means 50 µg of DNA per millilitres of water. The extinction coefficient for nucleic acid in water provides the basis for the relationship between absorbance and concentration.
Humic acid quantification is influenced by protein impurities and nucleic acid content. The number of humic acids, which is independent of the amount of DNA and protein, could be measured using absorbance at 320 nm.86 The degree of PCR contamination and A320 readings have a strong correlation (R2 = 0.911). The two methods of evaluating humic acid levels are (i) absorbance at 340 nm and (ii) fluorescence (excitation at 471 nm and emission at 529 nm).87 Humic acid tests utilizing either 10 ng/µl humic acid addition are affected by DNA and protein content. Humic acid concentrations and absorbance measurements at 340 nm range from 0.1 to 100 ng/µl. The humic compounds may be measured effectively using samples that contain 50 ng/µl of humic acid and either 10 ng/µl DNA or 2 µg/µl bovine serum albumin. These samples are identical to those that simply contain humic acid.
Metagenomic DNA library construction and functional screening
In order to screen the complex metagenomic libraries made up of millions of clones, a different function-driven approach that is highly selective for targeted genes is based on mutants of host strains or heterologous complementation of host strains that require the targeted genes for growth under the selective conditions. The identification of genes encoding lysine racemases,88 enzymes involved in poly-3-hydroxybutyrate metabolism,89 resistance to antibiotics,89-92 DNA polymerases, and lipase93 are among the functional screens using heterologous complementation. The creation of a metagenomic DNA library from diverse environmental samples and its subsequent cloning into an appropriate vector are contingent upon the quality of the extracted DNA (Figure 3). This is because the enzymatic modifications necessary for cloning are susceptible to contamination from a range of biotic and abiotic elements, including humic substances. Large chunks of DNA are needed if the material is to be utilized to build a gene bank, as this will reduce the number of clones that need to be screened. The fragment size of the DNA may not matter as much in a PCR as it does in terms of DNA yield. Building a metagenomic DNA library and effectively screening it are necessary steps towards the possible identification of new chemicals. When preparing a metagenomic DNA library, two factors need to be properly taken into consideration. The first is the size of the DNA fragments to be cloned from the metagenome, which is the entirety of the genomes of all the bacteria that have colonized a certain habitat. The genetic diversity of the soil metagenome is between 5,000 and 5,000,000 times higher than that of the E. coli genome, based on the reassociation kinetic data.4,94 The number and clustering of the genes involved in the production of new chemicals is the second factor to take into account. Because distinct taxonomic groups exist, prokaryotes exhibit large variances in expression, and random cloning in E. coli can only identify 40% of the enzymatic activity.95 To broaden the range of detectable activities in the functional screening of metagenomic DNA libraries, more hosts have been used, including Streptomyces spp.,96 Thermus thermophilus,97 Sulfolobus solfataricus,98 and diverse Proteobacteria.99 Numerous novel biocatalysts have been identified as a result of the use of metagenomic libraries. These include cellulases,21,100 DNA polymerases,101 proteases,24 lipases/esterases,30,102-104 and antibiotics (Table).90 The biocatalysts that have most commonly been retrieved from metagenomic libraries are likely lipases and esterases. A screening for b-D-glucuronidase activity was conducted on a metagenomic library consisting of 46,000 clones obtained from fecal pools.105 Metagenomic libraries should comprise clones that are commonly clustered and have bigger DNA inserts since the genes encoding the production of secondary metabolites are often clustered.
Table:
Functional screening for antibiotics, industrially important enzymes, and biocatalysts from soil metagenomic libraries
Gene |
Habitat |
Library type |
Average insert size (kb) |
Number of clones screened |
Substrate |
Positive Clones (hits) |
Ref |
|---|---|---|---|---|---|---|---|
Esterase/Lipase |
Soil |
Plasmid |
6 |
286,000 |
Tributyrin |
3 |
104 |
Esterase/Lipase |
Soil |
Plasmid |
6 |
730,000 |
Triolein |
1 |
104 |
Esterase/Lipase |
Soil |
BAC |
27 |
3,648 |
Bacto Lipid |
2 |
68 |
Esterase/Lipase |
Soil |
Plasmid |
8 |
1, 50,000 |
Tributyrin |
10 |
106 |
Esterase/Lipase |
Forest soil |
Fosmid |
40 |
31,000 |
Tributyrin |
7 |
107 |
Esterase/Lipase |
Soil |
Fosmid |
35 |
65,000 |
Tributyrin |
12 |
106 |
Esterase/Lipase |
Forest soil |
Fosmid |
35 |
33,700 |
Tributyrin |
8 |
108 |
Lipase |
Antarctic soil |
Plasmid |
5 |
1,000 |
Olive oil |
1 |
102 |
Lipase |
Forest soil |
Plasmid |
4.6 |
20,000 |
Tributyrin |
2 |
109 |
Amylase |
soil |
Plasmid |
5 |
80,000 |
Starch |
1 |
110 |
Amylase |
Soil |
BAC |
27 |
3,648 |
Starch |
1 |
67, 111, 112 |
Amylase |
Soil |
Fosmid |
35 |
76,000 |
Starch |
1 |
113 |
Amidase |
Soil |
Cosmid |
40 |
D-phenylglycine-L-leucine |
3 |
104 |
|
Amidase |
Soil |
Plasmid |
5 |
193,000 |
D-phenylglycine-L-leucine |
7 |
110, 114 |
Cellulase |
Sediment |
Λ phage |
6 |
310,000 |
Carboxymethyl cellulose |
3 |
115 |
Cellulase |
Soil |
Cosmid |
22 |
1700 |
Carboxymethyl cellulose |
8 |
20, 100 |
Protease |
Soil |
Plasmid |
10 |
1000,000 |
Skimmed milk |
1 |
116 |
Lipase |
Soil |
Plasmid |
2 |
87,000 |
Tributyrin |
1 |
98 |
4-Hydroxtbutyrate
Conversion |
Soil |
Plasmid |
6 |
930,000 |
4-Hydroxybutyrate |
5 |
117 |
Oxidoreductase |
Sediment |
Plasmid |
6 |
100000 |
Glycerol, 1,2-propanediol |
24 |
118 |
Dehydratase |
Soil |
Plasmid |
4 |
560,000 |
Glycerol |
2 |
119 |
Polyketide synthase (Type I) |
Soil |
Fosmid |
40 |
60000 |
Glycerol and chloramphenicol |
139 |
8 |
β-Lactamase |
Soil |
Plasmid |
5 |
80,000 |
Ampicillin |
4 |
110 |
PKSI |
Soil |
Cosmid |
– |
5,000 |
B. subtilis |
1 |
120 |
DNase-anti bacterial Comp.2 lipase |
Soil |
BAC |
27 |
3,648 |
nitrocefin |
81 |
67 |
DNase-anti bacterial Comp.2 lipase |
Soil |
BAC |
44.5 |
24576 |
nitrocefin |
132 |
67 |
Glycerol dehydratase Diol dehydratase |
Sediment |
Plasmid |
5 |
1,00000 |
Polyols, Carbonyls |
24 |
117 |
Polyketide synthase(Type I) |
Soil |
BAC |
120 |
– |
1 |
19 |
|
Long chain N-acyl amino acid antibiotic |
Soil |
Cosmid |
45 |
– |
B. subtilis |
11 |
121 |
Acyl tyrosine |
Soil |
Cosmid |
35 |
B. subtilis |
10 |
122 |
|
Bacteriorhodopsin |
Soil |
BAC |
80 |
2400 |
1 |
40, 41 |
|
Biotin |
Soil |
Cosmid |
35 |
50,000 |
Biotin-deficient medium |
7 |
123 |
Agarase |
Soil |
Cosmid |
38 |
1532 |
Agar medium |
18 |
28 |
Pectate lyase |
Soil |
Cosmid |
38 |
1532 |
Pectin |
2 |
28 |
Nitrilase |
Soil |
Λ phage |
10 |
109 |
Adiponitrile |
200 |
124 |
Turbomycin |
Soil |
BAC |
44.5 |
24,546 |
Several |
3 |
125 |
Indirubin |
Soil |
BAC |
63 |
12,000 |
B.subtilis, S.aureus |
4 |
126 |
Violacein, Deoxyviolacein |
Soil |
Cosmid |
40 |
– |
B.subtilis |
2 |
127 |
Terragine A |
Soil |
Plasmid |
2 |
1020 |
2 |
96 |
|
4-Hydroxybutyrate conversion |
Soil |
Plasmid |
6 |
930,000 |
4-Hydroxybutyrate |
5 |
116 |
Dehydratase |
Soil |
Plasmid |
4 |
560,000 |
Glycerol |
2 |
119 |
Alcohol Oxidoreductase |
Soil |
Plasmid |
4 |
400,000 |
Glycerol |
10 |
117 |
Cellulase |
Soil |
Illumina DNA library preparation kit |
3 |
– |
AZCL-Cellulose |
1 |
128 |
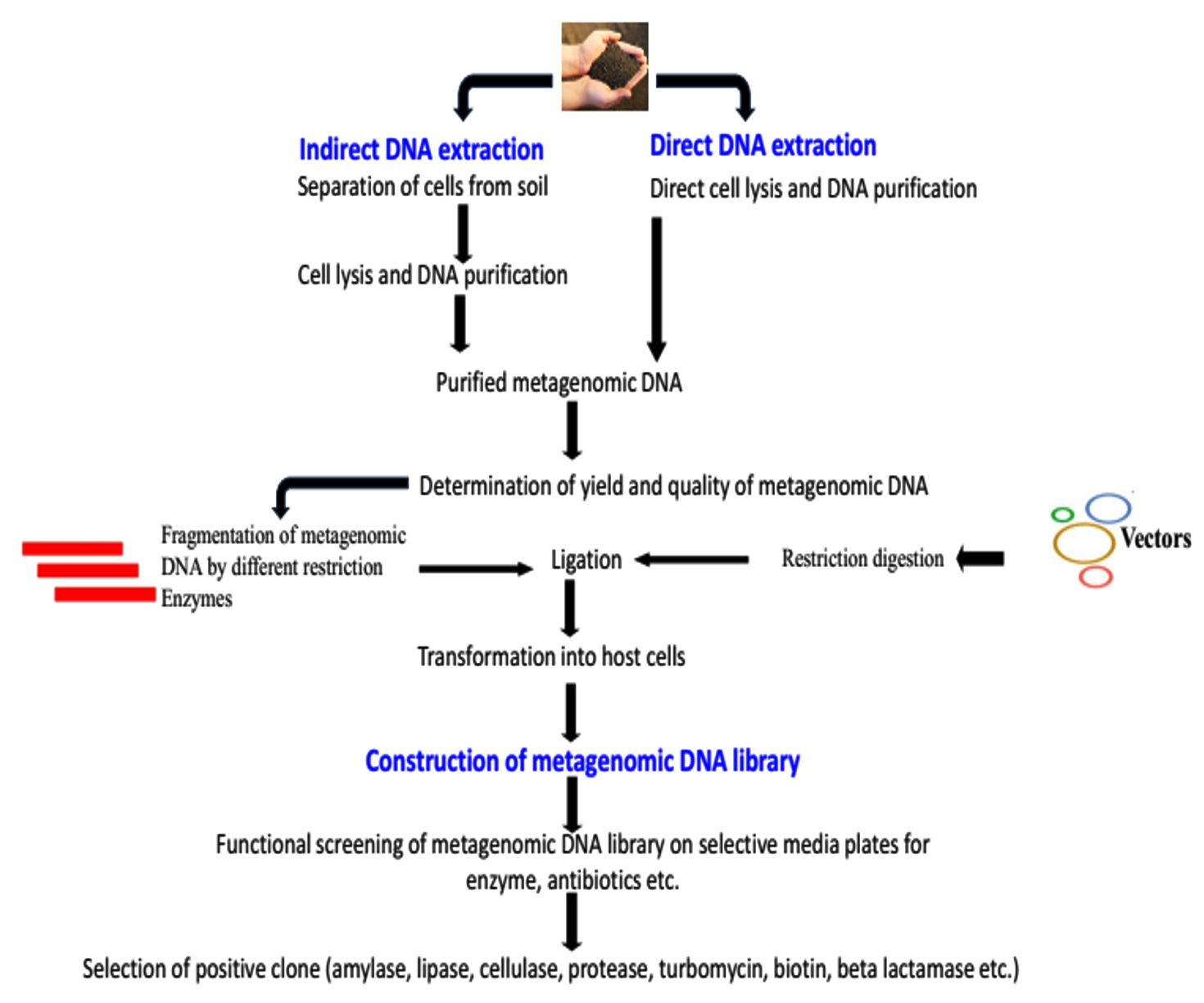
Figure 3. Workflow for the exploitation of gene through soil metagenomics
Bead beating and mechanical shearing pose physical challenges to direct DNA isolation techniques from soil and sediments for metagenomic library assembly.129, 130 DNA released during cell lysis may be deteriorated by nucleases. Given the frequent clustering of genes encoding the production of secondary metabolites, genomic libraries ought to include clones with bigger DNA inserts. Large portions of the genomes can be more easily characterized because to the cloning of high molecular weight DNA into bacterial artificial chromosomes (BACs).131 It is known that over 80% of the actinomycete promoters are not recognized by E. coli because of the variation in G+C composition.132 On the other hand, the maximum number of functionally expressed genes is required for functional screening. To isolate catabolic enzymes from the metagenome, a high throughput screening technology and substrate-induced gene expression screening have been used. When screening biocatalysts, the functional metagenomic approach is usually employed, which necessitates the expression of the required activity in a surrogate host, usually E. coli. A considerable fraction of proteins cannot be expressed in this host in a functional manner, despite the fact that the bacterial species has shown itself to be a versatile and helpful host for heterologous expression. The majority of functional experiments depend on the full expression of metabolic pathways, necessitating the identification of promoters in the heterologous host in order to coordinate the expression of all sets of genes. The entire potential of the microbial gene pool can be accessed using the metagenomic approach. Metagenomic clones that are expressed are extremely rare for any particular activity. For instance, out of 80,000, 730,000, and 100,000 clones that were found to have amylase, lipase, and protease activity, respectively, only one clone exhibited these activities. The clones were produced from soil. Three in 24,546 for turbomycin, four in 12,000 for indirubin, and seven in 50,000 for biotin synthesis were indicative of activity in a library built using soil metagenome. The development of effective functional screening and selection techniques is necessary due to the lack of functionally active clones, in order to find novel compounds such as medicines and industrially significant enzymes. The Daniel group developed a CVclever selection for Na+(Li+)/H+ antiporters.133,134 To enable growth on the medium containing 7.5 mM LiCl, the Daniel group must complement an E. coli mutant that is deficient in three Na+/H+ antiporters (nhaA, nhaB, and chaA). Using this method, two new antiporter proteins were found in a soil metagenomic collection of 14,80,000 clones. When the functions of interest don’t give a solid foundation for selection, high throughput screens can be used instead. The ability of metagenomic analysis to target genes and find new biotechnological applications is being enhanced by metagenomic libraries.135,136 Selection for antibiotic resistance resulted in the extraction of determinants resistant to aminoglycosides from soil,111 and tetracycline from human oral microbiota samples.137 A high-throughput screening method known as substrate-induced gene expression screening (SIGEX) combines fluorescence-activated cell sorting with an operon trap gfp expression vector. SIGEX is governed by regulatory elements that are situated in close proximity to catabolic genes and is predicated on the expression of catabolic genes. A metagenomic library obtained from groundwater revealed the presence of an aromatic-hydrocarbon-induced gene.17 The potential for effectors other than the particular substrates to activate transcriptional regulators is a disadvantage of this strategy. Product-induced gene expression (PIGEX) screening was another method that was introduced.138 Product information is identified by the expression of gfp included in PIGEX. In the presence of substrate benzamide, amidases from the metagenomic library made with activated sludge are screened using a benzoate-responsive transcriptional regulator called BenR.
The intriguing and relatively young field of metagenomics holds the promise of producing biologically active compounds and opening up new avenues for biotechnology to solve urgent global concerns. Only metagenomics can access the vast amount of information contained in the microbial genome. Since their inception, metagenome-based methods have resulted in the collection of a growing number of DNA sequences; nevertheless, up until now, the sequences that have been recovered are those of uncultivated bacteria. Humic acid contamination presents a considerable obstacle to soil DNA extraction and purification, therefore no one technology is suitable for this process. Combining various techniques may improve the production and calibres of DNA. It is now the time to accept modern techniques for the study of unexplored microorganism which are tough to culture in artificial media. Metagenomic technologies are projected to unlock millions of unique genes for drugs, pharmacological compounds, industrially significant enzymes for metabolic pathways, phylogenetic links, and microbial interactions among uncultured bacteria. Beyond revolutionizing mainstream microbiology, the field of metagenomics promises to revolutionize our understanding of the entire living universe through its fresh perspective on the microbial world.
ACKNOWLEDGMENTS
The authors are highly thankful to the Head of the Department of Botany and Microbiology for providing the necessary facilities.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
AUTHORS’ CONTRIBUTION
KS and HC conceptualized the study. KS, HC and KP wrote the manuscript. KKG, AK and AY reviewed and edited the manuscript. All authors read and approved the final manuscript for publication.
FUNDING
None.
DATA AVAILABILITY
All datasets generated or analyzed during this study are included in the manuscript.
ETHICS STATEMENT
Not applicable.
- Rajendhran J, Gunasekaran P. Strategies for accessing soil metagenome for desired applications. Biotechnol Adv. 2008;26(6):576-590.
Crossref - Turnbaugh PJ, Gordon JI. An invitation to the marriage of metagenomics and metabolomics. Cell. 2008;134(5):708-713.
Crossref - Sleator RD, Shortall C, Hill C. Metagenomics. Lett Appl Microbiol. 2008;47(5):361-366.
Crossref - Torsvik V, Daae FL, Sandaa RA, Ovreas L. Novel techniques for analysing microbial diversity in natural and perturbed environments. J Biotechnol.1998;64(1):53-62.
Crossref - Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian Classifier for Rapid Assignment of rRNA Sequences into the New Bacterial Taxonomy. Appl Environ Microbiol. 2007;73(16):5261-5267.
Crossref - Tringe SG, Von Mering C, Kobayashi A, et al. Comparative metagenomics of microbial communities. Science. 2005;308(5721):554-557.
Crossref - DeLong EF, Preston CM, Mincer T, et al. Community genomics among stratified microbial assemblages in the ocean’s interior. Science. 2006;311(5760):496-503.
Crossref - Handelsman J. Metagenomics: Application of genomics to uncultured microorganisms. Microbiol Mol Biol Rev. 2004;68(4):669-685.
Crossref - Biers EJ, Sun S, Howard EC. Prokaryotic genomes and Diversity in surface Ocean waters: Interrogating the Global Ocean Sampling Metagenome. Appl Environ Microbiol. 2009;75(7):2221-2229.
Crossref - Quince C, Walker AW, Simpson JT, Loman NJ, Segata N. Shotgun metagenomics, from sampling to analysis. Nat Biotechnol. 2017;35(9):833-844.
Crossref - Cowan D, Meyer Q, Stafford W, Muyanga S, Cameron R, Wittwer P. Metagenomic gene discovery: past, present and future. Trends Biotechnol. 2005;23(6):321-329.
Crossref - Daniel R. The soil metagenome – a rich resource for the discovery of novel natural products. Curr Opin Biotechnol. 2004;15(3):199-204.
Crossref - Tsai YL, Olson BH. Rapid method for separation of bacterial DNA from humic substances in sediments for polymerase chain reaction. Appl Environ Microbiol. 1992;58(7):2292-2295.
Crossref - Lemon KP, Klepac-Ceraj V, Schiffer HK, Brodie EL, Lynch SV, Kolter R. Comparative analyses of the bacterial microbiota of the human nostril and oropharynx. mBio. 2010;1(3):e00129.
Crossref - Wu C, Sun B. Identification of Novel Esterase from Metagenomic Library of Yangtze River. J Microbiol Biotechnol. 2008;19(2):187-193.
Crossref - Abulencia CB, Wyborski DL, Garcia JA, et al. Environmental Whole-Genome amplification to access microbial populations in contaminated sediments. Appl Environ Microbiol. 2006;72(5):3291-3301.
Crossref - Uchiyama T, Abe T, Ikemura T, Watanabe K. Substrate-induced gene-expression screening of environmental metagenome libraries for isolation of catabolic genes. Nat Biotechnol. 2004;23(1):88-93.
Crossref - Stein JL, Marsh TL, Wu KY, Shizuya H, DeLong EF. Characterization of uncultivated prokaryotes: isolation and analysis of a 40-kilobase-pair genome fragment from a planktonic marine archaeon. J Bacteriol. 1996;178(3):591-599.
Crossref - Ginolhac A, Jarrin C, Gillet B, et al. Phylogenetic analysis of polyketide synthase I domains from soil metagenomic libraries allows selection of promising clones. Appl Environ Microbiol. 2004; 70(9): 5522-5527.
https://doi.org/10.1128/AEM.70.9.5522-5527.2004″10.1128/AEM.70.9.5522-5527.2004 - Voget S, Steele HL, Streit WR. Characterization of a metagenome-derived halotolerant cellulase. J Biotechnol. 2006;126(1):26-36.
Crossref - Voget S, Leggewie C, Uesbeck A, Raasch C, Jaeger KE, Streit WR. Prospecting for novel biocatalysts in a soil metagenome. Appl Environ Microbiol. 2003;69(10):6235-6242.
Crossref - Hardeman F, Sjoling S. Metagenomic approach for the isolation of a novel low-temperature-active lipase from uncultured bacteria of marine sediment. FEMS Microbiol Ecol. 2006;59(2):524-534.
Crossref - Pathak GP, Ehrenreich A, Losi A, Streit WR, Gartner W. Novel blue light sensitive proteins from a metagenomic approach. Environ Microbiol. 2009;11(9):2388-2399.
Crossref - Waschkowitz T, Rockstroh S, Daniel R. Isolation and Characterization of Metalloproteases with a Novel Domain Structure by Construction and Screening of Metagenomic Libraries. Appl Environ Microbiol. 2009;75(8):2506-2516.
Crossref - Waschulin V, Borsetto C, James R, et al. Biosynthetic potential of uncultured Antarctic soil bacteria revealed through long-read metagenomic sequencing. ISME J. 2021;16(1):101-111.
Crossref - Hu X, Gu H, Liu J, et al. Metagenomics reveals divergent functional profiles of soil carbon and nitrogen cycling under long-term addition of chemical and organic fertilizers in the black soil region. Geoderma. 2022;418:115846.
Crossref - Rhee JK, Ahn DG, Kim YG, Oh JW. New Thermophilic and Thermostable Esterase with Sequence Similarity to the Hormone-Sensitive Lipase Family, Cloned from a Metagenomic Library. Appl Environ Microbiol. 2005;71(2):817-825.
Crossref - Simon C, Wiezer A, Strittmatter AW, Daniel R. Phylogenetic diversity and metabolic potential revealed in a glacier ice metagenome. Appl Environ Microbiol. 2009;75(23):7519-7526.
Crossref - Duan Cj, Xian L, Zhao Gc, et al. Isolation and partial characterization of novel genes encoding acidic cellulases from metagenomes of buffalo rumens. J Appl Microbiol. 2009;107(1):245-256.
Crossref - Heath C, Hu XP, Cary SC, Cowan D. Identification of a Novel Alkaliphilic Esterase Active at Low Temperatures by Screening a Metagenomic Library from Antarctic Desert Soil. Appl Environ Microbiol. 2009;75(13):4657-4659.
Crossref - Tyson GW, Chapman J, Hugenholtz P, et al. Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature. 2004;428(6978):37-43.
Crossref - Venter JC, Remington K, Heidelberg JF, et al. Environmental Genome shotgun sequencing of the Sargasso Sea. Science. 2004;304(5667):66-74.
Crossref - Zhang L, Chen F, Zeng Z, et al. Advances in metagenomics and its application in environmental microorganisms. Front Microbiol. 2021;12:766364.
Crossref - Arango-Argoty G, Garner E, Pruden A, Heath LS, Vikesland P, Zhang L. DeepARG: a deep learning approach for predicting antibiotic resistance genes from metagenomic data. Microbiome. 2018;6(1):23.
Crossref - Karkman A, DO TT, Walsh F, Virta MPJ. Antibiotic-Resistance genes in waste water. Trends Microbiol. 2017;26(3):220-228.
Crossref - Nnadozie CF, Odume ON. Freshwater environments as reservoirs of antibiotic-resistant bacteria and their role in the dissemination of antibiotic resistance genes. Environ Pollut. 2019;254(Pt B):113067.
Crossref - Diaz-Torres A, Thompson IJ, Beck C. How does breakup influence the total fusion of Li6,7 at the Coulomb barrier? Phys Rev C. 2003;68(4):044607.
Crossref - Ferrer M, Beloqui A, Timmis KN, Golyshin PN. Metagenomics for mining New genetic resources of microbial communities. Microb Physiol. 2008;16(1-2):109-123.
Crossref - Chistoserdova L. Recent progress and new challenges in metagenomics for biotechnology. Biotechnol Lett. 2010;32(10):1351-1359.
Crossref - Beja O, Suzuki MT, Koonin EV, et al. Construction and analysis of bacterial artificial chromosome libraries from a marine microbial assemblage. Environ Microbiol. 2000;2(5):516-529.
Crossref - Beja O, Spudich EN, Spudich JL, Leclerc M, DeLong EF. Proteorhodopsin phototrophy in the ocean. Nature. 2001;411(6839):786-789.
Crossref - Katara A, Chand S, Chaudhary H, Chandra H, Dubey RC. Microbial diversity of river Ganga at Haridwar (Uttarakhand) through metagenomic approaches. J Appl Biol Biotechnol. 2024;12(2):171556.
Crossref - Schneegurt MA, Dore SY, Kulpa CF Jr. Direct extraction of DNA from soils for studies in microbial ecology. Curr Issues Mol Biol. 2003;5(1):1-8.
- Yeates C, Gillings MR, Davison AD, Altavilla N, Veal DA. Methods for microbial DNA extraction from soil for PCR amplification. Biol Proc Online. 1998;1, 40-47.
Crossref - Holben WE, Jansson JK, Chelm BK, Tiedje JM. DNA probe method for the detection of specific microorganisms in the soil bacterial community. Appl Environ Microbiol. 1988;54(3):703-711.
Crossref - Singh SP, Sagar K and Konwar BK. Strategy in metagenomic DNA isolation and computational studies of humic acid. Curr Res Microbiol Biotechnol. 2013;1(1):9-11.
- Porteous LA, Seidler RJ, Watrud LS. An improved method for purifying DNA from soil for polymerase chain reaction amplification and molecular ecology applications. Mol Ecol. 1997;6(8):787-791.
Crossref - Borneman J, Skroch PW, O’Sullivan KM, et al. Molecular microbial diversity of an agricultural soil in Wisconsin. Appl Environ Microbiol. 1996;62(6):1935-1943.
Crossref - Clegg CD, Ritz K, Griffiths BS. Direct extraction of microbial community DNA from humified upland soils. Lett Appl Microbiol. 1997;25(1):30-33.
Crossref - Cullen DW, Hirsch PR. Simple and rapid method fordirect extraction of microbial DNA fromsoil for PCR. Soil Biol Biochem. 1998;30(8-9):983-993.
Crossref - Ramsay AJ. Extraction of bacteria from soil: Efficiency of shaking or ultrasonication as indicated by direct counts and autoradiography. Soil Biol Biochem. 1984;16(5):475-481.
Crossref - Picard C, Ponsonnet C, Paget E, Nesme X, Simonet P. Detection and enumeration of bacteria in soil by direct DNA extraction and polymerase chain reaction. Appl Environ Microbiol. 1992;58(9):2717-2722.
Crossref - Van Elsas JD, Mantynen V, Wolters AC. Soil DNA extraction and assessment of the fate of Mycobacterium chlorophenolicum strain PCP-1 in different soils by 16S ribosomal RNA gene sequence based most-probable-number PCR and immunofluorescence. Biol Fertil Soils. 1997;24(2):188-195.
Crossref - Miller DN, Bryant JE, Madsen EL, Ghiorse WC. Evaluation and optimization of DNA extraction and purification procedures for soil and sediment samples. Appl Environ Microbiol. 1999;65(11):4715-4724.
Crossref - Tsai YL, Olson BH. Rapid method for direct extraction of DNA from soil and sediments. Appl Environ Microbiol. 1991;57(4):1070-1074.
Crossref - Zhou J, Bruns MA, Tiedje JM. DNA recovery from soils of diverse composition. Appl Environ Microbiol. 1996;62(2):316-322.
Crossref - Frostegard ASA, Courtois S, Ramisse V, et al. Quantification of Bias Related to the Extraction of DNA Directly from Soils. Appl Environ Microbiol. 1999;65(12):5409-5420.
Crossref - Edgecombe GD, Giribet G, Wheeler WC. Phylogeny of Chilopoda: combining 18S and 28S rRNA sequences and morphology. Bol Soc Ent Aragonesa. 1999;(26):321-331.
- Ogram A, Sayler GS, Barkay T. The extraction and purification of microbial DNA from sediments. J Microbiol Methods. 1987;7(2-3):57-66.
Crossref - Lorenz MG, Wackernagel W. Adsorption of DNA to sand and variable degradation rates of adsorbed DNA. Appl Environ Microbiol. 1987;53(12):2948-2952.
Crossref - Weyant RS, Edmonds P, Swaminathan B. Effect of ionic and nonionic detergents on the Taq polymerase. Biotechniques. 1990;9(3):308-309.
- Roose-Amsaleg CL, Garnier-Sillam E, Harry M. Extraction and purification of microbial DNA from soil and sediment samples. Appl Soil Ecol. 2001;18(1):47-60.
Crossref - Gray JP, Herwig RP. Phylogenetic analysis of the bacterial communities in marine sediments. Appl Environ Microbiol. 1996;62(11):4049-4059.
Crossref - Selenska S, Klingmaller W. Direct detection of nif-gene sequences of Enterobacter agglomeransin soil. FEMS Microbiol Lett. 1991;80(2-3):243-245.
Crossref - Xia X, Bollinger J, Ogram A. Molecular genetic analysis of the response of three soil microbial communities to the application of 2, 4-D. Mol Ecol. 1995;4(1):17-28.
Crossref - Harry M, Gambier B, Bourezgui Y, Garnier-Sillam E. Evaluation of purification procedures for DNA extracted from rich organic samples: interference with humic substances. Analusis. 1999;27(5):439-441.
Crossref - Rondon MR, August PR, Bettermann AD, et al. Cloning the Soil Metagenome: a Strategy for Accessing the Genetic and Functional Diversity of Uncultured Microorganisms. Appl Environ Microbiol. 2000;66(6):2541-2547.
Crossref - Martin-Laurent F, Philippot L, Hallet S, et al. DNA Extraction from Soils: Old Bias for New Microbial Diversity Analysis Methods. Appl Environ Microbiol. 2001;67(5):2354-2359.
Crossref - Sagar K, Singh SP, Goutam KK, Konwar BK. Assessment of five soil DNA extraction methods and a rapid laboratory-developed method for quality soil DNA extraction for 16S rDNA-based amplification and library construction. J Microbiol Methods. 2013;97:68-73.
Crossref - Konwar BK, Sagar K. Lipase: An Industrial Enzyme Through Metagenomics (1st ed.). Apple Academic Press. 2018.
Crossref - Bouchez T, Blieux AL, Dequiedt S, et al. Molecular microbiology methods for environmental diagnosis. Environ Chem Lett. 2016;14(4):423-441.
Crossref - Nazir A. Review on metagenomics and its applications. Imp J Intersd Res. 2016 ;1;2(10).
- Piceno YM, Noble PA, Lovell CR. Spatial and temporal assessment of diazotroph assemblage composition in vegetated salt marsh sediments using denaturing gradient gel electrophoresis analysis. Microb Ecol. 1999;38(2):157-167.
Crossref - Herrick JB, Madsen EL, Batt CA, Ghiorse WC. Polymerase chain reaction amplification of naphthalene-catabolic and 16S rRNA gene sequences from indigenous sediment bacteria. Appl Environ Microbiol. 1993;59(3):687-694.
Crossref - Jackson CR, Harper JP, Willoughby D, Roden EE, Churchill PF. A simple, efficient method for the separation of humic substances and DNA from environmental samples. Appl Environ Microbiol. 1997;63(12):4993-4995.
Crossref - Edgcomb VP, Taylor C, Pachiadaki MG, Honjo S, Engstrom I, Yakimov M. Comparison of Niskin vs. in situ approaches for analysis of gene expression in deep Mediterranean Sea water samples. Deep Sea Research Part II Topical Studies in Oceanography. 2014;129:213-222.
Crossref - Manjula A, Sathyavathi S, Gunasekaran P, Rajendhran J. Comparison of seven methods of DNA extraction from termitarium for functional metagenomic DNA library construction. Sci Ind Res. 2011;(70):945-951.
- Plassart P, Terrat S, Thomson B, et al. Evaluation of the ISO Standard 11063 DNA extraction Procedure for assessing soil microbial abundance and community structure. PLoS ONE. 2012;7(9):e44279.
Crossref - Volossiouk T, Robb EJ, Nazar RN. Direct DNA extraction for PCR-mediated assays of soil organisms. Appl Environ Microbiol. 1995;61(11):3972-3976.
Crossref - Holben WE. Isolation and Purification of Bacterial DNA from Soil. In: Soil Science Society of America Book Series. 2013:727-751.
Crossref - Lovell CR, Piceno Y. Purification of DNA from estuarine sediments. J Microbiol Methods. 1994;20(3):161-174.
Crossref - Courtois S, Frostegard A, Garansson P, Depret G, Jeannin P, Simonet P. Quantification of bacterial subgroups in soil: comparison of DNA extracted directly from soil or from cells previously released by density gradient centrifugation. Environ Microbiol. 2001;3(7):431-439.
Crossref - Katara A, Chand S, Chaudhary H, Chaudhry V, Chandra H, Dubey RC. Evolution and applications of Next Generation Sequencing and its intricate relations with chromatographic and spectrometric techniques in modern day sciences. J Chromatogr Open. 2024;5:100121.
Crossref - Senesi N, Loffredo E. Soil humic substances. Biopolymers Online. 2001.
Crossref - Steffen KT, Hatakka A, Hofrichter M. Degradation of Humic Acids by the Litter-Decomposing Basidiomycete Collybia dryophila. Appl Environ Microbiol. 2002;68(7):3442-3448.
Crossref - Miller DN. Evaluation of gel filtration resins for the removal of PCR-inhibitory substances from soils and sediments. J Microbiol Methods. 2001;44(1):49-58.
Crossref - Howeler M, Ghiorse WC, Walker LP. A quantitative analysis of DNA extraction and purification from compost. J Microbiol Methods. 2003;54(1):37-45.
Crossref - Chen IC, Thiruvengadam V, Lin WD, Chang HH, Hsu WH. Lysine racemase: a novel non-antibiotic selectable marker for plant transformation. Plant Mol Biol. 2009;72(1-2):153-169.
Crossref - Wang C, Meek DJ, Panchal P, et al. Isolation of Poly-3-Hydroxybutyrate Metabolism Genes from Complex Microbial Communities by Phenotypic Complementation of Bacterial Mutants. Appl Environ Microbiol. 2006;72(1):384-391.
Crossref - Riesenfeld CS, Goodman RM, Handelsman J. Uncultured soil bacteria are a reservoir of new antibiotic resistance genes. Environ Microbiol. 2004;6(9):981-989.
Crossref - Denef VJ, VerBerkmoes NC, Shah MB, et al. Proteomics inferred genome typing (PIGT) demonstrates inter population recombination as a strategy for environmental adaptation. Environ Microbiol. 2008;11(2):313-325.
Crossref - Donato JJ, Moe LA, Converse BJ, et al. Metagenomic analysis of apple orchard soil reveals antibiotic resistance genes encoding predicted bifunctional proteins. Appl Environ Microbiol. 2010;76(13):4396-4401.
Crossref - Sagar K, Konwar BK. Construction of a metagenomic DNA library for a novel lipase from the bakery soil of Assam, India. Int J Res Anal Rev. 2020;7(1):148-151.
- Doolittle WF. Phylogenetic classification and the universal Tree. Science. 1999;284(5423):2124-2128.
Crossref - Gabor EM, Alkema WBL, Janssen DB. Quantifying the accessibility of the metagenome by random expression cloning techniques. Environ Microbiol. 2004;6(9):879-886.
Crossref - Wang GYS, Graziani E, Waters B, et al. Novel Natural Products from Soil DNA Libraries in a Streptomycete Host. Org Lett. 2000;2(16):2401-2404.
Crossref - Angelov A, Mientus M, Liebl S, Liebl W. A two-host fosmid system for functional screening of metagenomic libraries from extreme thermophiles. Syst Appl Microbiol. 2009;32(3):177-185.
Crossref - Albers Sv, Jonuscheit M, Dinkelaker S, et al. Production of Recombinant and Tagged Proteins in the Hyperthermophilic Archaeon Sulfolobus solfataricus. Appl Environ Microbiol. 2006;72(1):102-111.
Crossref - Craig JW, Chang FY, Kim JH, Obiajulu SC, Brady SF. Expanding Small-Molecule Functional Metagenomics through Parallel Screening of Broad-Host-Range Cosmid Environmental DNA Libraries in Diverse Proteobacteria. Appl Environ Microbiol. 2010;76(5):1633-1641.
Crossref - Healy FG, Ray RM, Aldrich HC, Wilkie AC, Ingram LO, Shanmugam KT. Direct isolation of functional genes encoding cellulases from the microbial consortia in a thermophilic, anaerobic digester maintained on lignocellulose. Appl Microbiol Biotechnol. 1995;43(4):667-674.
Crossref - Simon C, Herath J, Rockstroh S, Daniel R. Rapid Identification of Genes Encoding DNA Polymerases by Function-Based Screening of Metagenomic Libraries Derived from Glacial Ice. Appl Environ Microbiol. 2009;75(9):2964-2968.
Crossref - Kur J, Turkiewicz M. Identification and molecular modeling of a novel lipase from an Antarctic soil metagenomic library. Polish Society of Microbiologists. 2009;58(3):199-204.
- Elend C, Schmeisser C, Leggewie C, et al. Isolation and biochemical characterization of two novel Metagenome-Derived esterases. Appl Environ Microbiol. 2006;72(5):3637-3645.
Crossref - Henne A, Schmitz RA, Bomeke M, Gottschalk G, Daniel R. Screening of Environmental DNA Libraries for the Presence of Genes Conferring Lipolytic Activity on Escherichia coli. Appl Environ Microbiol. 2000;66(7):3113-3116.
Crossref - Gloux K, Berteau O, El-Oumami H, Beguet F, Leclerc M, Dore J. A metagenomic b-glucuronidase uncovers a core adaptive function of the human intestinal microbiome. Proc Natl Acad Sci. 2010;108(Suppl 1):4539-4546.
Crossref - de Castro AP, Quirino BF, Allen H, Williamson LL, Handelsman J, Kruger RH. Construction and validation of two metagenomic DNA libraries from Cerrado soil with high clay content. Biotechnol Lett. 2011;33(11):2169-2175.
Crossref - Hong KS, Lim HK, Chung EJ, et al. Selection and characterization of forest soil metagenome genes encoding lipolytic enzymes. J Microbiol Biotechnol. 2007;17(10):1655-1660.
- Lee SW, Won K, Lim HK, Kim JC, Choi GJ, Cho KY. Screening for novel lipolytic enzymes from uncultured soil microorganisms. Appl Microbiol Biotechnol. 2004;65(6):720-726.
Crossref - Jimenez DJ, Montana JS, Alvarez D, Baena S. A novel cold active esterase derived from Colombian high Andean-forest soil metagenome. World J Microbiol Biotechnol. 2011;28(1):361-370.
Crossref - Gabor EM. Harvesting novel biocatalysts from the metagenome. Thesis, Univ. Groningen. 2004.
- Riesenfeld CS, Schloss PD, Handelsman J. Metagenomics: Genomic analysis of microbial communities. Ann Rev Genet. 2004;38(1):525-552.
Crossref - Liles MR, Manske BF, Bintrim SB, Handelsman J, Goodman RM. A Census of rRNA Genes and Linked Genomic Sequences within a Soil Metagenomic Library. Appl Environ Microbiol. 2003;69(5):2684-2691.
Crossref - Vidya J, Swaroop S, Singh SK, Alex D, Sukumaran RK, Pandey A. Isolation and characterization of a novel a-amylase from a metagenomic library of Western Ghats of Kerala, India. Biologia. 2011;66(6):939-944.
Crossref - Gabor EM, De Vries EJ, Janssen DB. Construction, characterization, and use of small insert gene banks of DNA isolated from soil and enrichment cultures for the recovery of novel amidases. Environ Microbiol. 2004;6(9):948-958.
Crossref - Rees HC, Grant S, Jones B, Grant WD, Heaphy S. Detecting cellulase and esterase enzyme activities encoded by novel genes present in environmental DNA libraries. Extremophiles. 2003;7(5):415-421.
Crossref - Gupta R, Beg Q, Lorenz, P. Bacterial alkaline proteases: molecular approaches and industrial applications. Appl Microbiol Biotechnol. 2002;59(1):15-32.
Crossref - Henne A, Daniel R, Schmitz RA, Gottschalk G. Construction of Environmental DNA Libraries in Escherichia coli and Screening for the Presence of Genes Conferring Utilization of 4-Hydroxybutyrate. Appl Environ Microbiol. 1999;65(9):3901-3907.
Crossref - Knietsch A, Waschkowitz T, Bowien S, Henne A, Daniel R. Construction and Screening of Metagenomic Libraries Derived from Enrichment Cultures: Generation of a Gene Bank for Genes Conferring Alcohol Oxidoreductase Activity on Escherichia coli. Appl Environ Microbiol. 2003;69(3):1408-1416.
Crossref - Knietsch A, Bowien S, Whited G, Gottschalk G, Daniel R. Identification and Characterization of Coenzyme B12-Dependent Glycerol Dehydratase- and Diol Dehydratase-Encoding Genes from Metagenomic DNA Libraries Derived from Enrichment Cultures. Appl Environ Microbiol. 2003;69(6):3048-3060.
Crossref - Courtois S, Cappellano CM, Ball M, et al. Recombinant Environmental Libraries Provide Access to Microbial Diversity for Drug Discovery from Natural Products. Appl Environ Microbiol. 2003;69(1):49-55.
Crossref - Brady SF, Chao CJ, Clardy J. Long-Chain N-Acyltyrosine Synthases from Environmental DNA. Appl Environ Microbiol. 2004;70(11):6865-6870.
Crossref - Brady SF, Chao CJ, Handelsman J, Clardy J. Cloning and Heterologous Expression of a Natural Product Biosynthetic Gene Cluster from eDNA. Org Lett. 2001;3(13):1981-1984.
Crossref - Piel J, Hui D, Wen G, et al. Antitumor polyketide biosynthesis by an uncultivated bacterial symbiont of the marine sponge Theonella swinhoei. Proc Natl Acad Sci. 2004;101(46):16222-16227.
Crossref - Robertson DE, Chaplin JA, DeSantis G, et al. Exploring nitrilase sequence space for enantioselective catalysis. Appl Environ Microbiol. 2004;70(4):2429-2436.
Crossref - Gillespie DE, Brady SF, Bettermann AD, et al. Isolation of Antibiotics Turbomycin A and B from a Metagenomic Library of Soil Microbial DNA. Appl Environ Microbiol. 2002;68(9):4301-4306.
Crossref - MacNeil IA, Tiong CL, Minor C, August PR, et al. Expression and isolation of antimicrobial small molecules from soil DNA libraries. J Mol Microbiol Biotechnol.2001;3(2):301-308.
- Brady SF, Chao CJ, Clardy J. New natural product families from an environmental DNA (eDNA) gene cluster. J Am Chem Soc. 2002;124(34):9968-9969.
Crossref - Oliva B, Zervas A, Stougaard P, Westh P, Thogersen MS. Metagenomic exploration of cold active enzymes for detergent applications: Characterization of a novel, cold active and alkali stable GH8 endoglucanase from ikaite columns in SW Greenland. Microb Biotechnol. 2024;17(6):e14466.
Crossref - Smalla K, Cresswell N, Mendonca-Hagler LC, Wolters A, van Elsas JD. Rapid DNA extraction protocol from soil for polymerase chain reaction mediated amplification. J Appl Bacteriol. 1993;74(1):78-85.
Crossref - Yeates C, Gillings MR, Davison AD, Altavilla N, Veal DA. PCR amplification of crude microbial DNA extracted from soil. Lett Appl Microbiol. 1997;25(4):303-307.
Crossref - Berry AE, Chiocchini C, Selby T, Sosio M, Wellington EMH. Isolation of high molecular weight DNA from soil for cloning into BAC vectors. FEMS Microbiol Lett. 2003;223(1):15-20.
Crossref - Strohl WR. Compilation and analysis of DNA sequences associated with apparent streptomycete promoters. Nucleic Acids Res. 1992;20(5):961-974.
Crossref - MajerniK A, Gottschalk G, Daniel R. Screening of Environmental DNA Libraries for the Presence of Genes Conferring Na+(Li+)/H+Antiporter Activity on Escherichia coli: Characterization of the Recovered Genes and the Corresponding Gene Products. J Bacteriol. 2001;183(22):6645-6653.
Crossref - Ma YC, Auge RM, Dong C, Cheng ZM. Increased salt tolerance with overexpression of cation/proton antiporter 1 genes: a meta analysis. Plant Biotechnol J. 2016;15(2):162-173.
Crossref - Apolinar-Hernandez MM, Pena-Ramirez YJ, Perez-Rueda E, Canto-Canche BB, De Los Santos-Briones C, O’Connor-Sanchez A. Identification and in silico characterization of two novel genes encoding peptidases S8 found by functional screening in a metagenomic library of Yucatan underground water. Gene. 2016;593(1):154-161.
Crossref - Popovic A, Hai T, Tchigvintsev A, et al. Activity screening of environmental metagenomic libraries reveals novel carboxylesterase families. Sci Rep.2017;7(1):1-15.
Crossref - Diaz-Torres ML, Villedieu A, Hunt N, et al. Determining the antibiotic resistance potential of the indigenous oral microbiota of humans using a metagenomic approach. FEMS Microbiol Lett. 2006;258(2):257-262.
Crossref - Uchiyama T, Miyazaki K. Product-Induced Gene Expression, a Product-Responsive Reporter Assay used to screen metagenomic libraries for Enzyme-Encoding genes. Appl Environ Microbiol. 2010;76(21):7029-7035.
Crossref
© The Author(s) 2024. Open Access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License which permits unrestricted use, sharing, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.