


ISSN: 0973-7510
E-ISSN: 2581-690X
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the strain of virus that causes coronavirus disease 2019 (COVID-19), the respiratory illness responsible for the current pandemic. Viral genome sequencing has been widely applied during outbreaks to study the relatedness of this virus to other viruses, its transmission mode, pace, evolution and geographical spread, and also its adaptation to human hosts. To date, more than 90,000 SARS-CoV-2 genome sequences have been uploaded to the GISAID database. The availability of sequencing data along with clinical and geographical data may be useful for epidemiological investigations. In this study, we aimed to analyse the genetic background of SARS-CoV-2 from patients in Indonesia by whole genome sequencing. We examined nine samples from COVID-19 patients with RT-PCR cycle threshold (Ct) of less than 25 using ARTIC Network protocols for Oxford Nanopore’s Gridi On sequencer. The analytical methods were based on the ARTIC multiplex PCR sequencing protocol for COVID-19. In this study, we found that several genetic variants within the nine COVID-19 patient samples. We identified a mutation at position 614 P323L mutation in the ORF1ab gene often found in our severe patient samples. The number of SNPs and their location within the SARS-CoV-2 genome seems to vary. This diversity might be responsible for the virulence of the virus and its clinical manifestation.
SARS-CoV-19, COVID-19, RNA, Whole genome, SNP, Banten, Indonesia
The pandemic caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a global health issue. The first infection with this novel RNA coronavirus was reported in Wuhan, China, at the end of 2019, and since then it has spread throughout the world1,2,3. In Indonesia, the first case was identified in early March 2020, with reported cases of foreign nationals suffering from COVID-19. Gradually, the incidence of cases increased, especially in large cities4,5,6.
At the end of October 2020, COVID-19 cases in Indonesia had reached more than 350,000, with an average of nearly 4000 new cases per day. However, information about the SARS-CoV 2 RNA virus in Indonesia is still scant7,8.
As a medical faculty with a laboratory for SARS-CoV-2 examination, we have the opportunity to perform genomic analysis of SARS-CoV-2 samples from patients who have unfavourable clinical presentations. Using Minion Nanopore technology, we identified the genome of the SARS-CoV-2 from nine patients in Banten Province.
Sample collection
Since June 2020, The Medical Research Laboratory, Faculty of Medicine, Syarif Hidayatullah State Islamic University as become part of the COVID-19 Laboratory Examination Network for Banten, West Java, Indonesia. On average, we have received 400–500 samples every week, amounting to about 4000 samples by the end of October 2020, with a positivity rate of 25% (1000/4000 samples). We received samples from hospitals, clinics and public health centres in the Banten area to us in viral transport medium (VTM).In this study, we collected and sequenced nine RNA samples from these patients.
Ethical clearance
Ethical clearance was issued by the Ethic Committee of the Faculty of Medicine UIN Syarif Hidayatullah Jakarta (No.B-005/F12/KPK/TL.00/02/2021) and each specimen was submitted to our laboratory along with informed consent from the patient.
RNA extraction
Two hundred µL of VTM containing patient samples were transferred into a tube containing 600 µL of TRI-Zol solution (TRI Reagent®) and shaken until homogeneous. The homogeneous samples were centrifuged and the supernatants transferred into RNase-free tubes. Each supernatant in its RNase-free tube was mixed with 600 µL of ethanol (95–100%) and gently shaken. The mixture was transferred to a Zymo-SpinTM IC column in a collection tube, centrifuged, and the collection liquid discharged. Subsequently, 400 µL of RNA wash buffer was added into the column and centrifuged. This was followed by 5 µL of DNase I and 35 µL of DNA digestion buffer being added into the column matrix and incubated for 15 minutes at room temperature (RT). Then, 400 µL of Direct-ZolTM RNA prewash was added, centrifuged and the liquid flowing into the collection tube removed. Next,700 µL of RNA wash buffer was added to the column and centrifuged for two minutes to ensure that the wash buffer had transferred into the collection tube and the lysate had passed through the membrane. After that, the column was transferred into an RNase-free tube and 50 µL of DNase/RNase-free water was added to elute the RNA, followed by centrifugation so that the RNA flowed into the RNase-free tube.
RNA dissolved by 50 µL DNase/RNase-free water was stored at -80°C if not used immediately.
qRT-PCR analysis
We conducted qRT-PCR by using one step reaction Biosensor Standard M SARS-CoV-2 PCR (Chungcheongbuk-do, Republic of Korea) in a Roche LC480 II machine. The protocol was as described in the manufacture’s insert kit with targets of ORF1ab gene and E gene.
SARS-Cov-2 sequencing
Out of 6000 samples, we randomly selected nine samples from patients who were positive for COVID-19 and had a cycle threshold (Ct) value of between 15 and 25. All samples were measured for concentration and purity prior to sequencing.
All samples were prepared using ARTIC Network protocols and analytic methods9. The protocols and analyses were based on the ARTIC multiplex PCR sequencing protocol for COVID-19 devised by Josh Quick. The protocol generates 400 bp amplicons in a tiled fashion across the whole COVID-19 genome.
Oxford Nanopore’s GridION sequencer was operated using MinKNOW version 20.06.9 and MinKNOW core version 4.0.3. High accuracy base-calling was conducted using Guppy10 version 4.0.11. All generated reads were assembled using EPI2ME Labs platform employing ARTIC workflow.
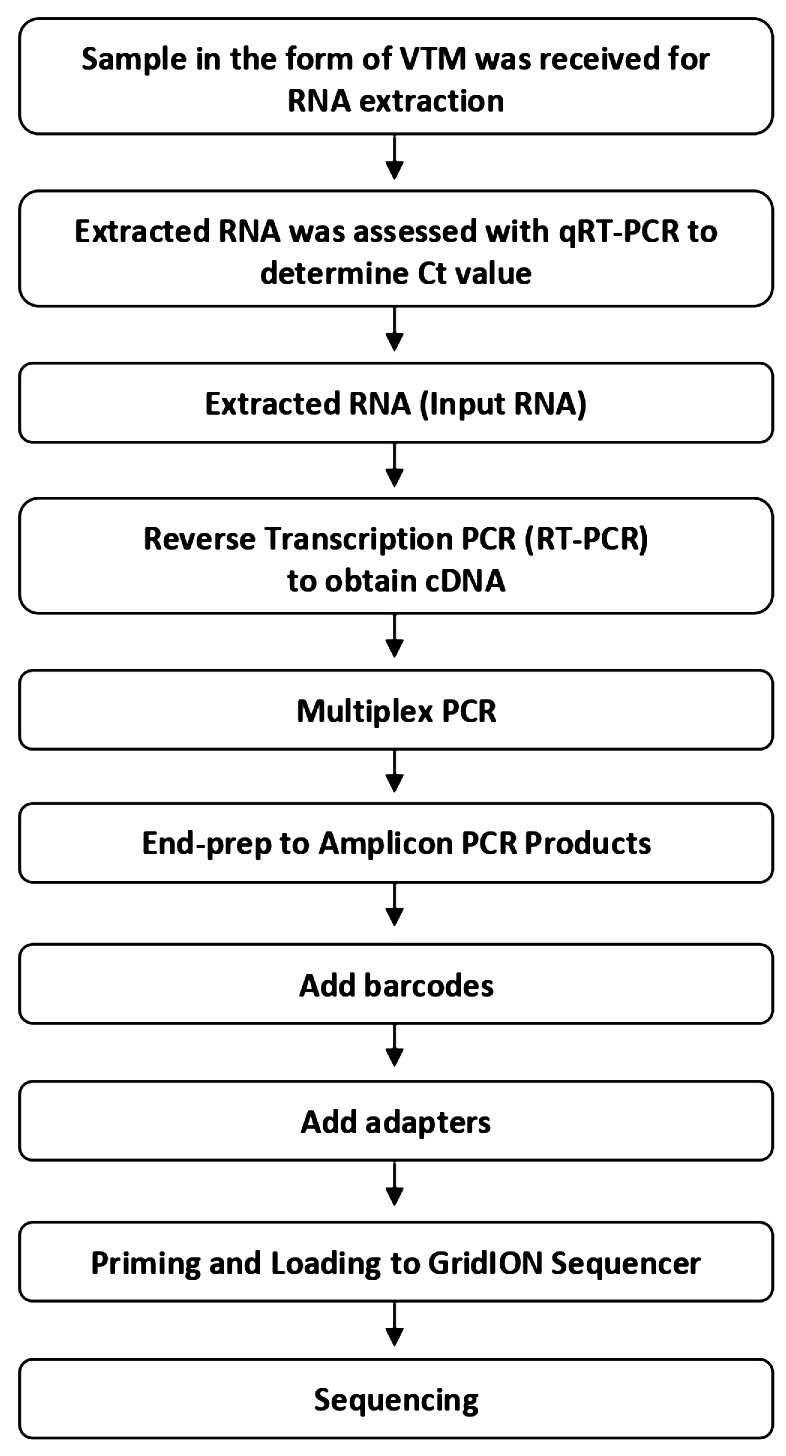
Fig. 1. Experimental workflow of sequencing
After preparation using ARTIC multiplex PCR, all samples were analysed using GridION operated by MinKNOW software. Base-calling was performed using Guppy with high accuration mode. Raw reads were assembled employing EPI2ME Labs software. All sequencing and bioinformatic workflows are shown in Figures 1 and 2.
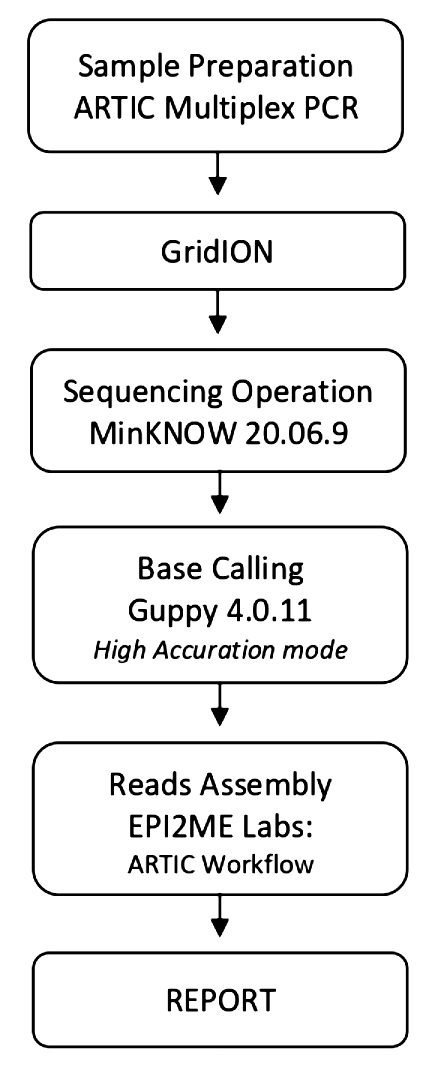
Fig. 2. Bioinformatic workflow9,10
In Indonesia, the number of COVID-19 patients reached more than 500,000 by the end of November 20201–8,11. Their clinical presentations varied from asymptomatic to severe.
In this study, we examined the whole genome of SARS-CoV-2 from nine COVID-19 patients using Nanopore’s GridION sequencer. We found variations in the changes of nucleotide bases in the exon and intergenic virus regions. The clinical characteristics of these patients are summarized in Table 1. Most of them had fever, anosmia, dry cough, and fatigue and did not have comorbidities.
Table (1):
Symptom and Ct of PCR target gene.
| Virus Name GISAID | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
|---|---|---|---|---|---|---|---|---|---|
| Parameter | hCoV- 19/Indonesia/B T-SHSIU-01- 2566/2020 | hCoV- 19/Indonesia/B T-SHSIU-01- 3610/2020 | hCoV- 19/Indonesia/B T-SHSIU-01- 3884/2020 | hCoV- 19/Indonesia/B T-SHSIU-01- 4920/2020 | hCoV- 19/Indonesia/B T-SHSIU-01- 4941/2020 | hCoV- 19/Indonesia/B T-SHSIU-01- 6368/2021 | hCoV- 19/Indonesia/B T-SHSIU-01- 6493/2021 | hCoV- 19/Indonesia/B T-SHSIU-01- 6494/2021 | hCoV- 19/Indonesia/B T-SHSIU-01- 6547/2021 |
| Clade | G | GH | GH | GH | GH | GR | GH | GH | GH |
| Lineage | B.1.459 | B.1.459 | B.1.468 | B.1.459 | B.1.459 | B.1.1.74 | B.1.470 | B.1.470 | B.1 |
| Gender | Male | Male | Male | Male | Female | Female | Female | Male | Female |
| Age ( in Year ) | 31 | 31 | 59 | 31 | 30 | 26 | 44 | 55 | 36 |
| Symptom | |||||||||
| Fever > 38 C | – | – | – | V | – | V | V | V | V |
| Dry Cough | – | V | – | V | – | V | V | V | V |
| Anosmia | – | – | – | V | – | V | V | V | V |
| Nausea | – | – | – | V | – | V | V | V | V |
| Vommit | – | V | – | – | – | V | V | V | V |
| Diarrhea | – | – | – | – | V | V | V | V | |
| Sore throat | – | – | – | V | – | V | V | V | V |
| Shorthness of breath | – | V | – | – | – | V | V | V | V |
| Fatigue | V | V | – | V | V | V | V | V | V |
| Malaise | – | V | – | – | – | V | V | V | V |
| Chest Radiography | – | V | – | – | – | – | – | – | – |
| Hypertension | – | – | – | – | – | – | – | – | – |
| Diabetes | – | – | – | – | – | – | – | – | – |
| Ct RT PCR Target Gene | |||||||||
| Orf1ab | 32 | 17 | 18 | 13 | 21 | 18 | 11 | 10 | 14 |
| E | 33 | 20 | 22 | 14 | 20 | 18 | 11 | 11 | 15 |
We found many missense and synonymous or silent mutations in the samples, as shown in Table 2. One sample was found to have a nucleotide deletion while the other changes were nucleotide substitutions. Moreover, we detected the gene area and amino acids that changed in these nine samples (Table 2) and most of the changes were found in the ORF1ab target gene. All of these SARS-CoV-2 sequences had already been submitted to GISAID.
The bioinformatic analysis using EPI2ME software revealed numbers of SNP variants ranging from 13 to 23 and one variant being a deletion.
As shown in Table 2, the most common nucleotide changes in our nine patients were C>T at nucleotide positions 241; 3037; 14408; 26735 and A>G at nucleotide position 23403. The codon and amino acid changes found in most samples were CCT>CTT.P>L proline to leucine, and GAT> GGT.D>G aspartate to glycine.
Table (2):
Type and region of mutation and nucleotide changes of SARS-CoV-2.
Virus Name GISAID |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
|---|---|---|---|---|---|---|---|---|---|
Parameter (Amino acid mutation) |
hCoV- 19/Indonesia/B T-SHSIU-01- 2566/2020 |
hCoV- 19/Indonesia/B T-SHSIU-01- 3610/2020 |
hCoV- 19/Indonesia/B T-SHSIU-01- 3884/2020 |
hCoV- 19/Indonesia/B T-SHSIU-01- 4920/2020 |
hCoV- 19/Indonesia/B T-SHSIU-01- 4941/2020 |
hCoV- 19/Indonesia/B T-SHSIU-01- 6368/2021 |
hCoV- 19/Indonesia/B T-SHSIU-01- 6493/2021 |
hCoV- 19/Indonesia/B T-SHSIU-01- 6494/2021 |
hCoV- 19/Indonesia/B T-SHSIU-01- 6547/2021 |
83 IR Start ORF1ab n.83T>G |
– |
V |
– |
– |
– |
– |
– |
– |
– |
241 IR Start ORF1ab n.241 C>T |
V |
V |
V |
V |
V |
V |
V |
V |
V |
1003 MV ORF1ab c.198A>C Glu66Asp |
– |
– |
– |
– |
– |
V |
– |
– |
– |
1568 MV ORF1ab c.763C>T His255Tyr |
– |
– |
– |
– |
– |
V |
– |
– |
– |
2158 SV ORF1ab c.1353T>A Leu451Leu |
– |
– |
– |
– |
– |
V |
– |
– |
– |
2273 MV ORF1ab c.1468G>A Glu490Lys |
– |
– |
– |
V |
– |
– |
– |
– |
– |
2891 MV ORF1ab c172G>A Ala58Thr |
– |
– |
V |
– |
– |
– |
– |
– |
– |
3037 SV ORF1ab c.318C >T Phe106Phe |
V |
V |
V |
V |
V |
V |
V |
V |
V |
3097 SV ORF1ab c.378A>G Ser126Ser |
– |
V |
– |
– |
– |
– |
– |
– |
– |
3239 MV ORF1ab c.2974G>T Asp174Ty |
– |
– |
– |
– |
V |
– |
– |
– |
– |
3768 MV ORF1ab c.1094C>T Thr350Ile |
– |
– |
– |
– |
– |
– |
– |
– |
V |
4084 SV ORF1ab c.1365C>T Asp455Asp |
V |
– |
– |
V |
– |
– |
– |
– |
– |
4543 SV ORF1ab c.1824C>T Thr608Thr |
– |
– |
– |
– |
V |
– |
– |
– |
– |
4754 MV ORF1ab c.2035C>T Pro679Ser |
– |
V |
– |
– |
– |
– |
– |
– |
– |
5184 MV ORF1ab c.2465C>T Pro822Leu |
V |
– |
– |
V |
V |
– |
– |
– |
V |
5806 SV ORF1ab c.3087C>T Cys1029Cys |
– |
– |
– |
– |
– |
V |
– |
– |
– |
6449 MV ORF1ab c.3730C>T Leu1244Phe |
– |
– |
V |
– |
– |
– |
– |
– |
– |
6675 MV ORF1ab c.3956C>T Thr1319Ile |
V |
– |
– |
– |
– |
– |
– |
– |
– |
7471 SV ORF1ab c.4752C>T Asp1584Asp |
– |
– |
V |
– |
– |
– |
– |
– |
– |
9711 SV ORF1ab c.1157C>T Ser386Phe |
– |
– |
– |
V |
– |
– |
– |
– |
– |
9970 SV ORF1ab c.1416A>T Ala472Ala |
– |
– |
– |
– |
– |
– |
– |
– |
V |
10030 SV ORF1ab c.1476C>T Thr492Thr |
– |
– |
– |
– |
– |
V |
– |
– |
– |
10089 MV ORF1ab c.35A>G Lys12ARG |
V |
– |
– |
V |
– |
– |
– |
– |
– |
10277 MV Orf1ab c.223C>T Leu75The |
– |
– |
– |
– |
– |
– |
V |
V |
– |
10507 SV ORF1ab c.453C>T Asn151Asn |
V |
– |
– |
V |
V |
– |
– |
– |
– |
11280 MV ORF1ab c.308C>A Thr103Asn |
– |
– |
– |
– |
V |
– |
– |
– |
– |
12754 SV ORF1ab c.69C>T Cys23Cys |
– |
– |
– |
– |
– |
– |
V |
V |
– |
13046 MV ORF1ab c.22C>T Pro8Ser |
– |
– |
– |
V |
– |
– |
– |
– |
– |
14120 MV ORF1ab c.680C>T Pro227Leu |
– |
– |
– |
– |
– |
– |
V |
V |
– |
14183 MV ORF1ab c.743C>T Thr248Ile |
V |
– |
– |
V |
– |
– |
– |
– |
– |
14408 MV ORF1ab c.968C>T Pro323Leu |
V |
V |
V |
V |
V |
V |
V |
V |
V |
14694 SV ORF1ab c.1254C>T Asp418Asp |
– |
V |
– |
– |
– |
– |
– |
– |
– |
14831 MV ORF1ab c.1391G>T Cys464Phe |
– |
V |
– |
– |
– |
– |
– |
– |
– |
15406 MV ORF1ab c.1966G>T Ala656Ser |
– |
V |
– |
– |
– |
– |
– |
– |
– |
15746 MV ORF1ab c.2306C>T Thr769Ile |
– |
– |
– |
– |
V |
– |
– |
– |
– |
16647 SV ORF1ab c.411G>T Thr137Thr |
V |
– |
– |
V |
– |
– |
– |
– |
– |
17004 SV ORF1ab c.768C>T Leu256Leu |
– |
– |
– |
– |
– |
– |
V |
V |
– |
17439 SV ORF1ab c.1203C>T Asp401Asp |
– |
– |
– |
– |
V |
– |
– |
– |
– |
17964 MV ORF1ab c.1728G>T Met576Ile |
– |
V |
– |
– |
– |
– |
– |
– |
– |
18169 MV ORF1ab c.130G>T Gly44Cys |
– |
– |
– |
– |
– |
– |
V |
V |
– |
18744 SV ORF1ab c.705C>T Tyr235Tyr |
– |
– |
– |
V |
V |
– |
– |
– |
– |
18877 SV ORF1ab c.838C>T Leu280Leu |
– |
V |
V |
V |
V |
– |
– |
– |
V |
19983 SV ORF1ab c.363C>T Val121Val |
V |
– |
– |
– |
V |
– |
– |
– |
– |
20320 MV ORF1ab c.700C>T His234Tyr |
– |
– |
– |
– |
– |
– |
– |
– |
V |
20389 MV ORF1ab c.769C>T Arg257Cys |
– |
– |
– |
– |
– |
– |
– |
– |
V |
21137 MV ORF1ab c.479A>G Lys160Arg |
– |
– |
– |
– |
– |
– |
V |
V |
– |
21575 MV S c.13C>T Leu5Phe |
– |
– |
– |
– |
V |
– |
– |
– |
– |
21595 SV S c.33C>T Val11Val |
– |
– |
– |
– |
V |
– |
– |
– |
– |
21742 SV S c.180C>T Ser60Ser |
– |
– |
– |
V |
– |
– |
– |
– |
– |
22021 MV S c.459G>T Met153Ile |
– |
– |
– |
– |
– |
V |
– |
– |
– |
22200 MV S c.638T>C Val213Ala |
V |
– |
– |
V |
– |
– |
– |
– |
– |
22487 MV S c.925G>C Glu309Gln |
– |
– |
– |
– |
– |
V |
– |
– |
– |
22879 MV S c.1317C>A Asn439Lys |
– |
– |
– |
– |
– |
– |
– |
– |
V |
23403 MV S c.1841A>G Asp614Gly |
V |
V |
V |
V |
V |
V |
V |
V |
V |
23593 MV S c.2031G>T Gln677His |
– |
– |
– |
– |
– |
– |
V |
V |
– |
24358 SV S c.2796 C>A Gly932Gly |
– |
– |
– |
– |
– |
– |
V |
V |
– |
25563 MV ORF3a c.171G>T Gln57His |
– |
V |
V |
V |
V |
– |
– |
– |
V |
25687 MV ORF3a c.295G>T Ala99Ser |
– |
V |
– |
– |
– |
– |
– |
– |
– |
25889 MV ORF3a c.497C>T Ser166Leu |
– |
– |
V |
– |
– |
– |
– |
– |
– |
25906 MV ORF3a c.514G>T Gly172Cys |
– |
– |
– |
– |
V |
– |
V |
V |
– |
26735 SV M c.213C>T Tyr71Tyr |
V |
V |
V |
V |
V |
– |
V |
V |
V |
26867 SV M c.345A>G Glu115Glu |
– |
– |
– |
V |
– |
– |
– |
– |
– |
27675 MV ORF7a c.282A>T Gln94His |
– |
– |
– |
– |
V |
– |
– |
– |
– |
28077 MV N c.184G>T Val62Leu |
– |
– |
V |
– |
– |
– |
– |
– |
– |
28628 MV N c.355G>T Ala119Ser |
– |
– |
– |
V |
– |
– |
– |
– |
– |
28752 MV N c.479A>G Gln160Arg |
– |
V |
– |
– |
– |
– |
– |
– |
– |
28851 MV N c.578G>T Ser193Ile |
V |
– |
– |
V |
– |
– |
– |
– |
– |
28880 MV N c.607A>G Arg203Gly |
– |
– |
– |
– |
– |
– |
V |
V |
– |
28881 MV N c.608G>T Arg203Met |
– |
– |
– |
– |
V |
– |
– |
– |
– |
28881 MV N c.608_610delGGGinsAAC ArgGly203LysArg |
– |
– |
– |
– |
– |
V |
– |
– |
– |
28887 MV N c.614C>T Thr205Ile |
– |
– |
– |
– |
– |
– |
– |
– |
V |
28985 MV N c.712G>T Gly238Cys |
– |
– |
V |
– |
– |
– |
– |
– |
– |
29095 SV N c.822C>T Phe274Phe |
– |
V |
– |
– |
– |
– |
– |
– |
– |
29749 IR ORF10-Ed n.29750_29751delCG |
– |
– |
V |
– |
– |
– |
– |
– |
– |
It has been reported that the SARS-CoV-2 genome is similar to that of the SARS-CoV virus that caused the epidemic in 2003. Overall, the protein characteristics for SARS-CoV have been identified, consisting of polyproteins Orf1a and Orf1ab; four structural proteins spike (S), envelope (E), membrane protein(M), and nucleocapsid (N); and eight accessory protein forms: Orf3a, Orf3b, Orf6, Orf7a, Orf7b, Orf8a, Orf8b, and Orf9. Accessory proteins, besides their function in viral replication, play a role in the interaction of the virus with its host.
In SARS-CoV-2,there are 11 proteins: Orf1ab, Orf2 (referred to as S protein); Orf3a and Orf4 are E proteins; Orf5 is the M protein; Orf6, Orf7a, Orf7b, Orf8, and Orf9 are N proteins, and finally Orf1012–17. In SARS-CoV-2, the Orf1ab gene expresses polyproteins, consisting of 16 non-structural proteins (NSP). NSP1 is known as an inhibitor of host gene expression binding to the host’s 40S ribosome which results in selective degradation of the host’s mRNA so that the viral mRNA can bind18,19. NSP2 has the ability to influence the host cell environment by binding to prohibitin proteins (PHB) 1 and 2 of host cells and resulting in cell cycle progression, cell migration, cellular differentiation, apoptosis and mitochondrial biogenesis20. NSP3 is a protease protein that plays a role in the release of essential viral proteins. The interaction of NSP3 and NSP4 is very important for viral replication21,22.
Mutations in viruses aim to adapt to the environment and new hosts. The capacity of viruses to adapt to their new hosts and environments depends to a great extent on their ability to produce diversity in a short period of time. Thus, the rate of spontaneous mutation between viruses varies widely due to the diversity-producing element encoded by the virus and its host. Viral diversity can also occur in response to certain selective stresses. RNA viruses have the ability to mutate faster than DNA viruses. Understanding the rate of virus mutation has implications for treatment, the development of drug resistance, immunity, pathogenesis and vaccination in efforts to control the disease23,24.
The most common types of SNPs detected in this study are missense and synonymous variants in the exon area. Missense mutations in the exon can affect codon changes for amino acids in the translation process. The effects of synonymous or silent mutations will affect post-transcriptional mRNA processes25–28. Such nucleotide mutations in the exon region enable changes in the amino acid sequence and alterations of the tertiary structure of the target protein.
Moreover, the intergenic region is an area where transcriptional enhancer sites related to regulatory functions are often found. Mutations in this area will affect the regulatory processes of the gene29,30–33. Therefore, all of these changes can result in the phenotypic diversity of the virus. It has been shown that mutations in the Orf area can affect the translation termination process34. Bali et al. demonstrated that synonymous mutations will affect protein function35.
Overall, missense mutation in the codon region was most prevalent in our samples. Changes due to missense will change the amino acid code and ultimately the amino acid that will be translated. Changes of proline (P) to leucine (L), and aspartate (D) to glycine(G) were those found in all of our samples.
Missense variants can affect the tertiary structure of a protein depending on the nature or character of the amino acids that form the polypeptide sequence. Changes of polar amino acids to non-polar, charged or neutral, will affect the properties of the proteins. We found 15 missense variants in the Orf1ab gene in our samples, and any change in amino acid residues will affect the structure of the Orf1ab polyprotein, which is known toplay a role in adaptation and virulence in its host. Graham et al. have described the possible role of Orf1ab in the pathogenic immune response of viruses and their hosts36. The Orf1ab polyprotein consists of 16 non-structural proteins (NSP)37. Non-structural proteins have a role in directing viral assembly after the virus invades the host cell, including viral transcription and replication, proteolytic processing, suppression of immune responses and expression of host genes37-41. One of our samples had an alteration in the amino acid residue glutamate to lysine at position 2273 of Orf1ab gene at amino acid position 670 (part of NSP3). The glutamate residue is a polar amino acid and the acid is replaced by an alkaline lysine residue. Changes in the sequence of amino acids in a polypeptide or protein in an organism can have a positive or negative effect on the organism, one of which is the ability of the virus to adapt to its host. NSP3 plays many roles in the viral life cycle as it can act as a scaffold protein to interact with itself and to bind to other viral NSPs or host proteins. NSP3 is also very important for the formation of replication transcription complex (RTC). RTC is linked to the host endoplasmic reticulum membrane to produce convoluted membranes and double membrane vesicles in SARS-CoV-2. Changes in the nucleotide bases or amino acids of NSP3 are likely to have an influence in the role of NSP342. However, comprehensive information regarding the association of Glu670Lys with the virulence of SARS-CoV-2 is needed.
The conversion of aspartate residue to glycine residue at position 614 (Asp614Gly) of the S gene was found in all of our samples. The S gene plays a role in the formation of the spike protein (S), such that the S protein will be recognized and will bind to the host receptor so that the SARS-CoV-2 virus can enter the host cell. Several studies have reported a possible link between changes in D614G and the ability of the virus to infect host cells. This variant mutation was found at multiple geographic levels43–47. The aspartate residue at position 614 lies outside the receptor-binding domain or RBD and does not change the affinity of the S protein to bind ACE2, but is thought to play a role in ACE2-mediated cell transduction. In addition, it appears that S protein played a key role in the evolution of the coronavirus in circumventing the host’s immune mechanism43,47–49.
It is interesting that we also found another variant besides D614G in the S protein in two of our patients, namely Q677H50. This variant was first reported in Surabaya, East Java, and was also found in our patients living in Banten, West Java. Personal communication with patients provides information on the possibility of contact with relatives or family members who are from East Java. This shows the importance of lockdown measures during the SARS-CoV-2 pandemic to limit or prevent the widespread of highly virulent virus variants.
In addition to mutations in the S protein, we observed that patients with severe conditions had missense mutations in spikeD614G, in NS3(Orf3a)Q57H, and NSP12 (Orf1ab) P323L. Majumdar et al. report an association between mutations in Orf3a and manifestations of SARS-CoV-2 immuno-pathogenic infection51. Wu et al. report that mutations in Q57H cause a dramatic change in protein structure that would affect the binding affinity for antiviral proteins52. The ORF3a protein is one of the largest accessory proteins in SARS-CoV-2 and is a link in the pathogenesis of COVID-1953. The mutation in the NSP12 as RdRp catalyses the replication of RNA and could affect the speed of viral replication54. The combination of three mutations in this region could lead to more severe clinical presentation and fatality of COVID-19.
Mutations are expected as natural events within the viral life cycle. Viral adaptation to the host usually results in higher transmission potential, as has been observed for SARS-CoV, MERS, and influenza. The pattern and time course of mutations in virus genomes are critical in estimating phylogenetic trees, which, in turn, depict the epidemic course effectively in real time. Mutations can provide information for understanding emerging outbreaks. The field of genomic epidemiology is presently employed in the mitigation and control of the SARS-CoV-2 outbreak55.
This study has shown several changes in the nucleotide sequence of SARS-CoV-2 resulting in different variants. The changes that occur cause the virus to survive and may lead to detrimental impact on the host. Therefore, molecular characterization and epidemiological data for SARS-CoV-2 variations are needed to monitor population shifts and to promptly identify emerging and more virulent variants. More importantly, vaccine efficacy against SARS-CoV-2 variants should be verified.
ACKNOWLEDGMENTS
The authors acknowledge their institutes and universities. We are grateful to Prof. HJ Freisleben for his valuable comments on this manuscript and Genetics Science for assisting in lending Nanopore Minions so that we could carry out this research.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
AUTHORS’ CONTRIBUTION
All authors listed have made substantial, direct and intellectual contributions to the work, and approved it for publication.
FUNDING
None.
ETHICS STATEMENT
Not applicable.
AVAILABILITY OF DATA
All data set generated or analyzed during this study are included in the manuscript.
- Mackenzie JS, Smith DW. Covid-19: a novel zoonotic disease caused by a coronavirus from China: what we know and what we don’t. Microbiol Aust. 2020;MA20013.
Crossref - Zheng J. SARS-CoV-2: an emerging Coronavirus that causes a global treat. Int J Biol Sci. 2020;16(10):1678-1685.
Crossref - Hu B, Guo H, Zhou P, Shi Z. Characteristic of SARS-CoV-2 and Covid-19. Nat Rev Microbiol. 2021;19:141-154.
Crossref - Ariawan I, Jusril H. COVID-19 in Indonesia: Where are we?. Acta Med Indones. 2020;52(3):193-195. PMID: 33020329
- Van Empel G, Mulyanto J, Wiratama BS. Undertesting of COVID-19 in Indonesia: what has gone wrong? J Glob Health. 2020;10(2):020306.
Crossref - Halim DA, Kurniawan A, Agung FA, et al. Understanding of young people about COVID-19 during early outbreak in Indonesia. Asia Pac J Public Health. 2020;32(6-7):363-365.
Crossref - Covid-19 di Indonesia: Kasus tembus 500.000 (Covid-19 in Indonesia: Cases penetrating 500,000, experts say the spread of the virus is’ increasingly undetected, the real number could be millions) https://www.bbc.com/indonesia/indonesia-55046484
- COVID-19 in Indonesia on October 31, 2020. Report of the Ministry of Health of the Republic of Indonesia. https://www.kemkes.go.id/resources/download/info-terkini/covid%20dalam%20angka/covid%20dalam%20angka%20-%2031102020.pdf
- Tyson JR, James P, Stoddart D, et al. Improvements to the ARTIC multiplex PCR method for SARS-CoV-2 genome sequencing using nanopore. BioRxiv. 2020.
Crossref - Wick RR, Judd LM, Holt KE. Performance of neural network basecalling tools for Oxford Nanopore sequencing. Genome Biol. 2019;20: 129.
Crossref - Positive COVID-19 Cases in Indonesia Add 6,267 People, 3T and 3M Efforts continue to be intensified. Report of the Ministry of Health of the Republic of Indonesia. https://www.kemkes.go.id/article/view/20113000001/kasus-positif-covid-19-di-indonesia-tambah-6-267-orang-upaya-3t-dan-3m-terus-digencarkan.html
- Laha S, Chakraborty J, Das S, Manna SK, Biswas S, Chatterjee R. Characterizations of SARS-CoV-2 mutational profile, spike protein stability and viral transmission. Infect Genet Evol. 2020;85:104445.
Crossref - Naqvi AAT, Fatima K, Mohammad T, et al. Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: Structural genomics approach. Biochim Biophys Acta Mol Basis Dis. 2020;866(10):165878.
Crossref - YA Helmy, M Fawzy, A Elaswad, A Sobieh, SP Kenney, AA Shehata. The COVID-19 pandemic: a comprehensive review of taxonomy, genetics, epidemiology, diagnosis, treatment and control. J Cli Med. 2020;9(4):1225.
Crossref - Khailany RA, Safdar M, Ozaslan M. Genomic characterization of a novel SARS-CoV-2. Gene Rep. 2020;19:100682.
Crossref - Abdelrahman Z, Li M, Wang X. Comperative review of SARS-CoV-2, SARS-CoV, MERS-CoV and influenza A respiratory viruses. Front Immuno. 2020;11:552909.
Crossref - Kaur M, Sharma A, Kumar S, Singh G, Barnwal RP. SARS-CoV-2: Insights into its structural intricacies and functional aspects for drug and vaccine development. Int J Biol Macromol. 2021;15(179):45-60.
Crossref - Schubert K, Karousis ED, Jomaa A, et al. SARS-CoV-2 Nsp1 binds the ribosomal mRNA channel to inhibit translation. Nat Struct Mol Biol. 2020;27(10):959-966.
Crossref - Lapointe CP, Grosely R, Johnson AG, Wang J, Fernandez IS, Puglisi JD. Dynamic competition between SARS-CoV-2 NSP1 and mRNA on the human ribosome inhibits translation initiation. Proc Natl Acad Sci USA. 2021;118(6):e2017715118.
Crossref - Cornillez-Ty CT, Liao L, Yates, JR, Kuhn P, Buchmeier MJ. Severe Acute Respiratory Syndrome Coronavirus NonStructural Protein 2 interacts with a host protein complex involved in mitochondrial biogenesis and intracellular signaling. J Virol. 2009; 83(19): 10314-10318.
Crossref - Sakai Y, Kawachi K, Terada Y, Omori H, Matsuura Y, Kamitani W. Two-amino acids change in the nsp4 of SARS coronavirus abolishes viral replication. Virology. 2017;510:165-174.
Crossref - Zhang J, Lan Y, Sanyal S. Membrane heist: Coronavirus host membrane remodeling during replication. Biochimie. 2020;179:229-236.
Crossref - Sanjuan R, Domingo-Calap P. Review: Mechanisms of viral mutation. Cell Mol Life Sci. 2016;73:4433-4448.
Crossref - Agudelo-Romero P, Carbonell P, Perez-Amador M, Elena SF. Virus adaptation by manipulating of host’s gene expression. Plos One.2008;3(6):e2397.
Crossref - Hunt RC, Simhadri VL, Iandoli M, Sauna ZE, Kimchi-Sarfaty C. Exposing synonymous mutations. Trends Genet. 2014;30(7):308-321.
Crossref - Sauna ZF, Kimchi-Sarfaty C, Ambudkar SV, Gottesman MM. The sounds of silence: synonymous mutations affect function. Pharmacogenomics. 2007;8(6):527-532.
Crossref - Komar AA. Silent SNPs: impact on gene function and phenotype. Pharmacogenomics. 2007;8(8):1075-1080.
Crossref - Shastry BS. SNPs: Impact on Gene Function and Phenotype. Method Mol Biol. 2009;578:3-22.
Crossref - Khademi SMH, Sazinas P, Jelsbak L. Within-Host Adaptation Mediated by Intergenic Evolution in Pseudomonas aeruginosa. Genome Biol Evol. 2019;11(5):1385-1397. PMID: 30980662.
Crossref - Macintrye G, Yepes AJ, Ong CS, Vespoor K. Associating disease-related genetic variants in intergenic regions to the genes they impact. Peer J. 2014;2:e639.
Crossref - Alderete JP, Jarrahian S, Geballe AP. Translational Effects of Mutations and Polymorphisms in a Repressive Upstream Open Reading Frame of the Human Cytomegalo virus UL4 Gene. J Virol. 1999;73(10):8330-8337. PMID: 10482583.
Crossref - Kumar P. Pandey R, Sharma P, et al. Integrated genomic view of SARS-CoV-2 in India. Wellcome Open Res. 2020;5:184.
Crossref - Li H, Achour A, Bastarache L, et al. Integrative genomics analysi analyses unveil downstream biological effectors of disease-specific polymorphisms buried in intergenic regions. npj Gen Med. 2016;1:16006.
Crossref - Schierding W, Antony J, Cutfield W, et al. Intergenic GWAS SNP are key components of spatial and regulatory network of human growth. Hum Mol Genet. 2016;25(15):3372-3382.
Crossref - Bali V, Bebok Z. Decoding Mechanisms by which Silent Codon Changes Influence Protein Biogenesis and Function. Int J Biochem Cell Biol. 2015; 64: 58-74.
Crossref - Graham RL, Spark JS, Eckerie LD, Sims AC, Denison MR. SARS coronavirus replicase proteins in pathogenesis. Virus Res. 2008;133(1):88-100.
Crossref - Yoshimoto FK. The Proteins of Severe Acute Respiratory Syndrome Coronavirus-2 (SARS CoV-2 or n-COV19), the Cause of COVID-19. The Prot J. 2020;39(6):198-216.
Crossref - Da Silva SJR, da Silva CTA, Mendes RPG, Pena L. Role of nonstructural proteins in the pathogenesis of SARS-CoV-2. J Med Virol. 2020;92(9)1427-1429.
Crossref - Banerjee AK, Blanco MR, Bruce EA, et al. SARS-CoV-2 Disrupts Splicing, Translation, and Protein Trafficking to Suppress Host Defenses. Cell. 2020;185(5):1325-1339.
Crossref - Snijder EJ, Decroly, Ziebuhr J. The nonstructural proteins directing coronavirus RNA synthesis and processing. Adv Virus Res. 2016;96:59-126.
Crossref - Denison MR, Graham RL, Donaldson EF, Eckerle LD, Barid RS. Coronaviruses: an RNA proofreading machine regulates replication fidelity and diversity. RNA Biol. 2011;8(2):270-279.
Crossref - Lei J, Kusov Y, Hilgenfeld R. Nsp3 of coronaviruses: Structures and functions of a large multi-domain protein. Antiviral Res. 2018;149:58-74.
Crossref - Volz E, Hill V, McCrone JT, et al. Evaluating the Effects of SARS-CoV-2 Spike Mutation D614G on Transmissibility and Pathogenicity. Cell. 2021;184(1):64-75.
Crossref - Korber B, Fischer WM, Gnanakaran S, et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell. 2020;182(4):812-827.
Crossref - Zhang L, Jackson CB, Mou H, et al. The D614G mutation in the SARS-CoV-2 spike protein reduces S1 shedding and increases infectivity. BioRxiv. 2020.
Crossref - Zhou B, Thao TTN, Hoffmann D, et al. SARS-CoV-2 spike D614G variant confers enhanced replication and transmissibility. BioRxiv. 2020.
Crossref - Groves DC, Rowland-Jones SL, Angyal A. The D614G mutations in the SARS-CoV-2 spike protein: Implications for viral infectivity, disease severity and vaccine design. Biochem Biophys Res Commun. 2021;538:104-107.
Crossref - Begum J, Mir NA, Dev K, Buyamayum B, Wani MY, Raza M. Challenges and prospects of COVID-19 vaccine development based on the progress made in SARS and MERS vaccine development. Transbound Emerg Dis. 2021;68(3):1111-1124.
Crossref - Isabel S, Grana-Miraglia L, Gutierrez J, et al. Evolutionary and structural analyses of SARS-CoV-2 D614G spike protein mutation now documented worldwide. Sci Rep. 2020;10(1):14031.
Crossref - Ansori ANM, Kharisma V D, Muttaqin SS, Antonius Y, Parikesit AA. Genetic variant of SARS-CoV-2 isolates in Indonesia: Spike glycoprotein gene. J Pure Appl Microbiol. 2020;14(Suppl1):971-978.
Crossref - Majumdar P, Niyogi S. ORF3a mutation associated with higher mortality rate in SARS-CoV-2 infection. Epidemiol Infect. 2020;148:e262.
Crossref - Wu S, Tian C, Liu P, et al. Effects of SARS-CoV-2 mutations on protein structures and intraviral protein-protein interactions. J Med Virol. 2021;93(4):2132-2140.
Crossref - Hassan SS, Choudhury PP,Basu P, Jana SS. Molecular conservation and differential mutation on ORF3a gene in Indian SARS-CoV2 genomes. Genomics. 2020;112(5):3226-3237.
Crossref - Vilar S, Isom DG. One year of SARS-CoV-2: How much has the virus changed? Biology. 2021;10:91.
Crossref - Grubaugh ND, Petrone ME, Holmes CE. We shouldn’t worry when a virus mutates during disease outbreaks. Nature Microbiology. 2020;5;529-530.
Crossref
© The Author(s) 2021. Open Access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License which permits unrestricted use, sharing, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.