


ISSN: 0973-7510
E-ISSN: 2581-690X
Oxalic acids are widely distributed in tissues of various plants that can exacerbate the effect on other plant-grazing animals including humans. Bacterial communities had been demonstrated to specify with the rhizosphere of host type and which can differ with oxalogenic plant. The present study has been conducted with the primary objective of understanding the root-associated microbial communities in Colocasia esculenta, an oxalogenic plant and to recognize possible bacterial species that present the potential of having their capability to metabolize oxalates. Of the 852 sequences obtained, 311 corresponded to rhizosphere (S), 250 to rhizoplane (P) and 291 were from non-rhizospheric (NS) soil. Flavobacteriaceae, Enterobacteriaceae, Moraxellaceae and Pseudomonadaceae were the major contributors in the rhizoplane microbial community assemblage. Paenibacillaceae was the major contributor to the rhizospheric microbial community. The findings of the study showed that the rhizoplane, owing to the characteristic root exudates, has a distinctive composition of microbial partners as compared to the rhizosphere and bulk soil communities.
Colocasia esculenta, rhizosphere soil, microbial community.
Oxalate (ethane-1,2-dioic acid) is found in diverse environments such as soil and gastrointestinal tracts; oxalate metabolizing bacteria commonly known as oxalotrophic bacteria that can metabolize oxalate for carbon and energy source are often enriched in such environments. In fact, only bacteria have the ability to degrade oxalate and remove it from the environments. Rhizosphere of the the oxalogenic plants is one such niche for such oxalotrophic bacteria.1,2. Recent literature has demonstrated that oxalotrophy phenomenon is involved in root colonization by plant-associated bacterial species like Burkholderia3 that may have a positive role in plant growth.
Oxalates, especially calcium oxalate (CaOx) crystals have been recorded in many plant species4 that can accumulate in plant-eating insects and animals including humans through a complex food network. Most of the times, some plant produces these oxalate crystals as a defense against the herbivory. However, the ingestion of oxalates can be toxic to mammals and may lead to hyperoxaluria and urolithiasis5. In fact, plants release oxalate in surrounding soil via root exudates6 and also contribute to the CaOx pool in the soil during the decay process7. The global oxalotrophic bacterial diversity has not been fully explored, diverse habitats can be explored with the aim of isolating and cultivating oxalotrophic bacteria. These oxalotrophic bacteria could then be utilized in the health and agriculture sectors to cope up with the increasing oxalate concentrations. Colocasia esculenta commonly known as arum has been documented to have oxalogenic properties8 and the present study has been conducted with the primary objective of understanding the rhizospheric microbial communities in an oxalogenic plant with the prospects of recognizing possible bacterial species for their capability to metabolize oxalates.
Plant and sampling
Naturally growing Colocasia esculenta (Local name: Arum) plants were collected in sterile polypropylene containers from a botanical garden near National Centre for Cell Science, situated in Savitribai Phule Pune University campus, Pune, Maharashtra, India (Latitude: 18°34′ N, Longitude: 73°58′ E). The plant roots and surrounded area were selected for sampling the non-rhizospheric (NS), rhizospheric (S) and rhizoplane (P) soil fractions. Soil from roots was fractionated into the three compartments following the previously described method9. Samples were selected after the randomization where the soil samples from six to eight arum plants were pooled, resulting in three independent biological replicates. The rhizosphere sample was collected by the gentle scraping of the portion with soil particles closely adhering to the root surface by using a sterile spatula and, root system without soil particles was considered as the rhizoplane sample. DNA from all the three fractions was isolated using PowerSoil® DNA isolation kit (MO BIO Laboratories, Inc. Carlsbad, CA) as per the manufacturer’s protocol provided.
Denaturing Gradient Gel Electrophoresis and qPCR for 16S rRNA gene
The fingerprint pattern of the all tested sample was generated by 16S rRNA gene PCR-DGGE and qPCR assay described elsewhere10,11. Briefly, PCR-DGGE was carried out on each community DNA with modified linker primers: 341F-GC (5’-CGCCCGCCGCGCGCGGCGGGCGGG GCGGGGGCACGGGGGGC CTACGGGAGGCA GCAG-3’) and 518R (5’-ATTACCGCGGCTGCTGG-3’) for total diversity10,12.
Quantitative estimation of the total bacterial population is achieved through 16S rRNA gene target and which was done in a triplicate using qPCR assays as described earlier 11. Average values of the triplicate of the single sample were used for enumerations of 16S rRNA gene copy numbers. Absolute count was derived for each sample using standard curves after the manual curation. All the quantitative values were scaled-up and represented in per gram of soil sample.
16S rRNA gene amplification and sequence data processing
16S rRNA gene was amplified in triplicate for each sample by polymerase chain reaction using bacteria-specific primers (8F: 5′-AGAGTTTGATCCTGGCTCAG-3′ and 907R: 5′-CCGTCAATTCCTTTRAGTTT-3′). Purified PCR products were then ligated to TOPO 2.1 TA vector using TOPO 2.1 TA cloning kit (Invitrogen, USA) and the vector containing 16S copies were then transformed into electrocompetent E. coli cells. Nearly 400 transformants were selected and sequenced using ABI Prism 3730 XL DNA Analyser (Applied BioSystems Inc., USA) as described earlier13. Sequences were assembled and quality checked using ChromasPro v1.34 (http://www.technelysium.com.au/ChromasPro.html), checked for orientation using orientation checker 14 and aligned in Clustal X2 15. Chimeric sequences were removed using Mallard v1.0210 and anomalous sequences removed using Pintail 16. A total of 852 good quality sequences were analyzed using QIIME-Quantitative Insight into Microbial Ecology v1.9 17. Operational Taxonomic Units (OTUs) were picked using an open reference method with a uclust_ref tool 18 using default 97% similarity value, followed by picking the most abundant sequence as the OTU representative sequence against the SILVA123 database (12 10-release)19.
Taxonomic assignments to each generated sequence
We utilized two independent approaches to assign the taxonomy to each sequence after the OTU generation. In the first approach, each representative OTU was utilized for taxonomical classification using a SILVA123 database with 97% identity as a reference. In an alternative approach, the same representative OTU file were utilized for taxonomical classification through BLAST using curated type strain 16S rRNA gene database on the EzTaxon-e server (http://www.ezbiocloud.net)20. For every OTU, the first hit was selected with 97% similarity cut-off and taxonomically identified at the species level. These identified OTUs were used for assessing the differences in the bacterial population between the three samples.
PICRUSt analysis
The functional metagenome imputation was done using the bioinformatic method as described earlier [21]. Briefly, OTUs from a closed reference-based OTU picking approach were utilized to bin the quality sequences. We used the Greengenes database v.13.5 for at 97% sequence similarity level cut-off to taxonomic profiling. The biom table utilized for PICRUSt analysis with available parameters at http://huttenhower.sph.harvard.edu/galaxy/. As steps mentioned, the OTU table was normalized for the 16S rRNA gene copy number. Next to that ascertains to for predicting metagenome with the help of the KEGG database at three different KEGG levels (L1 to L3). Additionally, prediction of oxalate bioconversion pathways present in imputed metagenome was made using KEGG Orthology (KO) database22 as mentioned elsewhere10.
Statistical analysis
Statistical analysis was done as mentioned elsewhere10. Briefly, statistical inferences were done in the STAMP-Software package for Taxonomic and Metabolic Profiling v2.08 23. Taxonomic profiling including genus-level taxonomy (L6) biom file containing all the relative abundance values, generated using QIIME command summarize taxa.py analyzed for qualitative ways using GraphPad Prism tool24. To compare 16S rRNA gene sequence libraries for the dispersion obtained from the three root compartments, here we treating each gene sequences as separate points in space with hundreds of dimensions, K-shuff, a statistical tool based on PHYLIP was used25. The diversity, species richness (OTUs) in the non-rhizosphere, rhizosphere and rhizoplane regions were analyzed by Simpson and Shannon indices along with goods coverage calculated by alpha diversity.py in QIIME environment and in DOTUR algorithm.
Nucleotide sequence accessions
Good quality 16S rRNA gene partial sequences of an average 850 base pair generated in this study which were deposited in Genbank-NCBI database with the following accession numbers: KF784899 – KF785750.
Microbial diversity
The fingerprinting pattern of the three root zones as analyzed through DGGE indicate a marked difference in the patterns. The actual bacterial count as obtained through qPCR indicates higher bacterial count in rhizosphere and lowest in rhizoplane as indicated by the following values: NS= 8.9 x 109, S= 6.1 x 1010 and P= 9.2 x 104 counts/gm of the soil sample. A total of 852 sequences were successfully retrieved from the three root compartments. Of these 852, 311 corresponded to rhizosphere (S), 250 to rhizoplane (P) and 291 were from non-rhizospheric (NS) soil. The 852 sequences obtained were clustered into representative 504 OTUs based on the open reference OTU clustering with 97% cutoff. This representative OTU file was used for BLAST analysis over EzTaxone-Server (Validly Culturable bacterial database), further resulting in 272 OTUs (97% cut off). Bacterial OTUs (271) were identified with species-level whereas 01 OTU corresponded to eukaryote. 148 sequences were identified and 160 unidentified at the species level in the rhizospheric soil. 208 sequences identified and 41 were unidentified at the species level in the rhizoplane soil. Similarly, in the non-rhizospheric soil, 273 sequences were identified and 17 rendered unidentified (Supplementary File S1).
The taxonomic assignment of each sequence to define OTUs was based on a similarity threshold of 97% to the SILVA database. Alpha diversity through non-parametric indicators: Simpson and Shannon were computed to evaluate community diversity characteristics of overall bacteria associated with the Colocasia esculenta roots (Fig. 1).
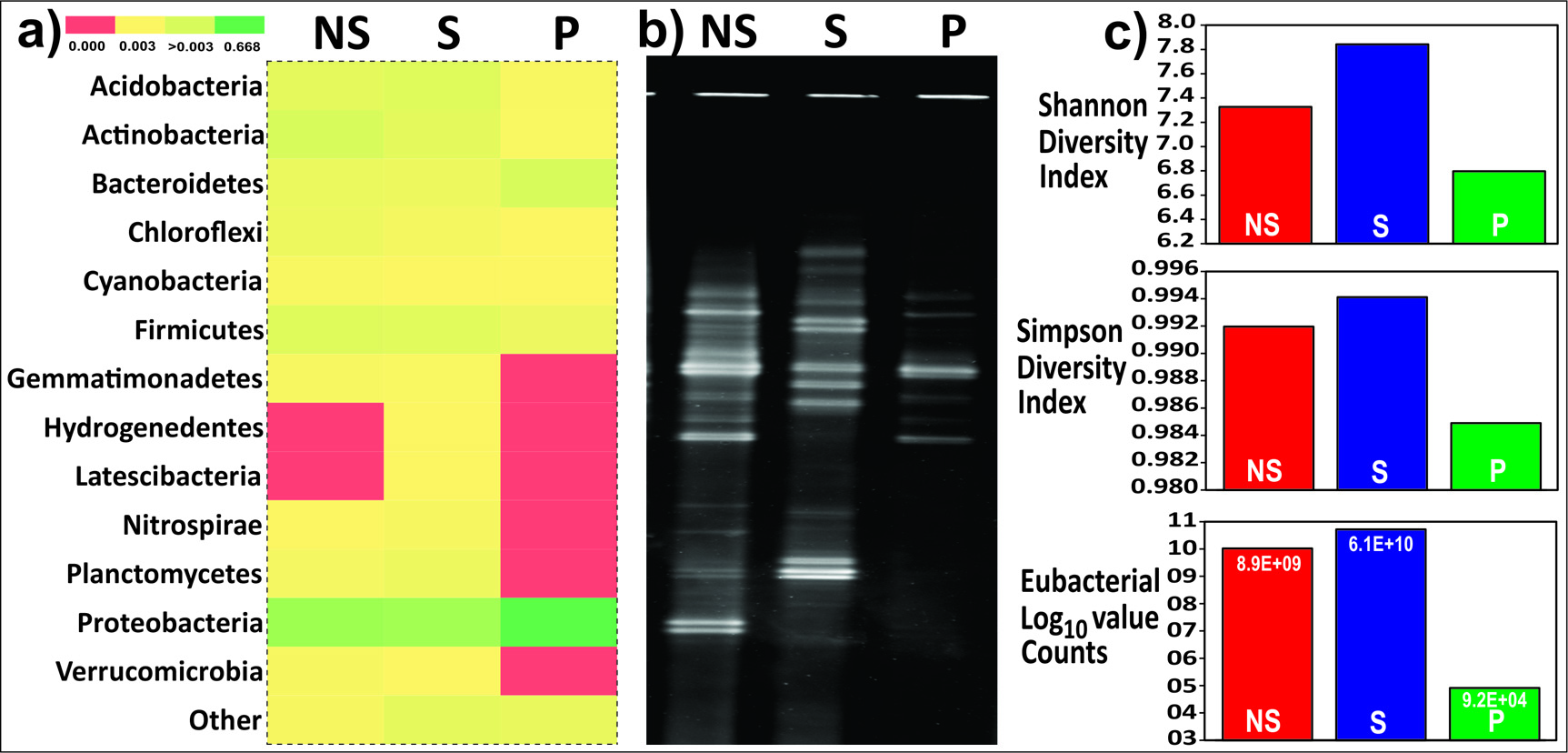
Fig. 1. Comparison of bacterial diversity in root-associated soil (Database used SILVA123) a) Heatmap for phylum-level diversity b) Fingerprint of root-associated microbiome by PCR-DGGE c) Graphical representation of alpha diversity indices and actual eubacteria count by quantitative PCR.
Bacterial community composition and structure
Taxonomic analysis based on relative abundance revealed bacterial members of phylum Proteobacteria were recorded relatively higher abundance across all samples S (48%), P (78%), and NS (40%). Firmicutes in rhizosphere (S) soil (28%), Actinobacteria (16%) in the non-rhizospheric soil whereas, Bacteroidetes (12%) in the rhizoplane soil were found to the second most abundant groups. The P and S showed a complete absence of members from the phyla Cyanobacteria, Gemmatemonadetes and Planctomycetes in contrast to the NS soil. Members from the phylum NC10 (0.68%), were exclusive to the rhizosphere soil. Acidobacteria, Chloroflexi, Nitrospirae, and Verrumicrobia were completely absent in the rhizoplane (P) soil samples (Fig. 1). K-shuff analysis showed a greater difference in microbial diversity of rhizoplane from bulk soil and rhizosphere as suggested by higher AreaK values generated (Table 1).
Table (1):
Eubacterial taxonomic features of root-associated diversity.
| Diversity Features | Clone library for microbiome study | ||
|---|---|---|---|
| NS | S | P | |
| Total Sequences (N) | 291 | 311 | 250 |
| dOTUs a | 172 | 228 | 60 |
| Shannon (H) b | 4.93049 | 5.1482 | 2.94576 |
| Simpson (D) c | 0.00578268 | 0.00780002 | 0.112161 |
| IKF d | 0.3049 | 0.2935 | 0.2554 |
| sOTUs e | 195 | 262 | 152 |
| Species level assignments f | 273 | 148 | 208 |
| Exclusion g | 1 | 3 | 1 |
| Newer to species level assignments h | 17 | 160 | 41 |
a Number of OTUs formed at 97 % sequence similarity using DOTUR (35).
b Shannon diversity index, H= -Σ[(n/N)ln(n/N)]; at Hmax, n= N.
c Simpson’s index, D= Σn(n-1)/N(N-1)
d IKF index derived from K Shuff algorithm (16).
e Number of OTUs formed at 97 % sequence similarity using the Silva123 database by open-reference OTU generation in OIIME environment (8).
f Number of library sequences at 97 % sequence similarity using EzTaxon database to the sOTUs.
g Number of library sequences having Eukaryotic taxonomy assignments.
h Number of library sequences fails to taxonomic assignments at 97 % sequence similarity using EzTaxon database to the sOTUs.
At the family level, the OTUs were characterized by 101 different families. Microbial communities from non-rhizospheric soil (NS) corresponded to 86 families. In contrast, the rhizoplanic (P) microbial communities corroborated to only 17 families. Flavobacteriaceae, Enterobacteriaceae, Moraxellaceae, and Pseudomonadaceae were the major contributors in the rhizoplane microbial community assemblage. Paenibacillaceae was the major contributor to the rhizospheric microbial community (Fig. 2).

Fig. 2. Heatmap for the family level abundance of root-associated soil microbiome (Database used SILVA123). Abundance in each library shown in the actual number and the clustering of each family-level taxa with dendrogram is derived with the help of farthest neighbour (constrained) method.
The bacterial species, particularly those belonging to the phylum Firmicutes, present among these communities are demonstrated through a Venn diagram to gauge the relationships between these communities (Fig. 3). The results illustrate that only 3 species were shared among all the three root zones. There were no species belonging to Firmicutes that were shared by non-rhizospheric (NS) and rhizoplane (P) soil samples. However, 6 bacterial species were shared by non-rhizospheric and rhizospheric soil samples. 6 bacterial species were exclusively present in the rhizoplane compartment which constituted of species belonging to genus Exiguobacterium, Paenibacillus, and Solibacillus.
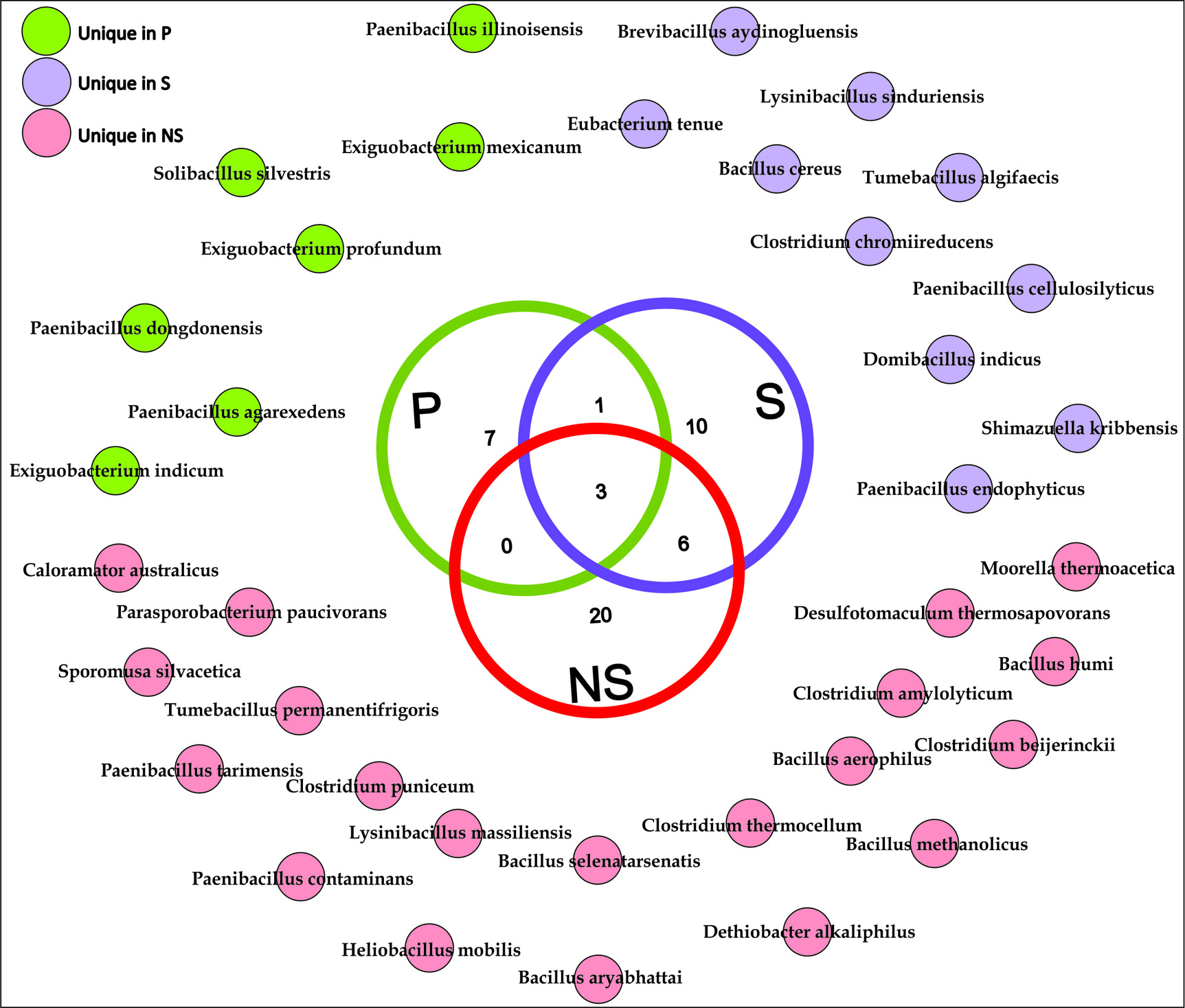
Fig. 3. Chart showing the sharing of all observed bacterial species of Firmicutes phylum for root-associated soil microbiome. These species-level taxonomic assignments were derived at 97 % sequence similarity using EzTaxon-e server.
Functional imputed metagenome
We compared the functional potential of the microbial communities in soil fractions (NS, P, and S), used bioinformatics tool- PICRUSt (Phylogenetic Investigation of Communities by Reconstruction of Unobserved States). Overall predicted genes that are more abundant in the rhizosphere were mainly associated with viz. Cellular Processes and Signalling, Cell Growth and Death, Carbohydrate Metabolism, Glycan Biosynthesis and Metabolism and Transcription (Fig. 4). Apart from other functional capabilities, other microbial metabolism enclosure was dominant (Supplementary File S2). PICRUSt was also employed to study the oxalate metabolism genes in the rhizosphere (Table 2) and bacterial families harboring oxalate metabolizing capacity genes are mentioned in Supplementary File S3.
Table (2):
Bacterial enzymes describing the oxalate metabolizing potentials in the microbiome derived from the imputed metagenomic approach. Selected oxalate metabolizing ability governing genes in microbiome as mentioned earlier by Suryavanshi et. al., 2016 13.
| Enzymes | KEGG Orthology ID | Abundance of genes in microbiome study | KEGG_Description | ||
|---|---|---|---|---|---|
| NS | S | P | |||
| oxalyl-CoA decarboxylase | K01577 | 0.0022 | 0.0016 | 0 | oxalyl-CoA decarboxylase [EC:4.1.1.8] |
| formyl-CoA transferase | K07749 | 0.0239 | 0.0219 | 0.0069 | formyl-CoA transferase [EC:2.8.3.16] |
| oxalate/formate antiporter | K08177 | 0.0107 | 0.0099 | 0.0122 | MFS transporter, OFA family, oxalate/formate antiporter |
| formyl-CoA hydrolase | K01067 | 0.0059 | 0.0059 | 0.0081 | acetyl-CoA hydrolase [EC:3.1.2.1] |
| NAD-dependent formate dehydrogenase | K08349 | 0.0003 | 0.0012 | 0.0106 | formate dehydrogenase-N, beta subunit [EC:1.2.1.2] |
| oxalate decarboxylase | K01569 | 0.0076 | 0.0040 | 0.0016 | oxalate decarboxylase [EC:4.1.1.2] |
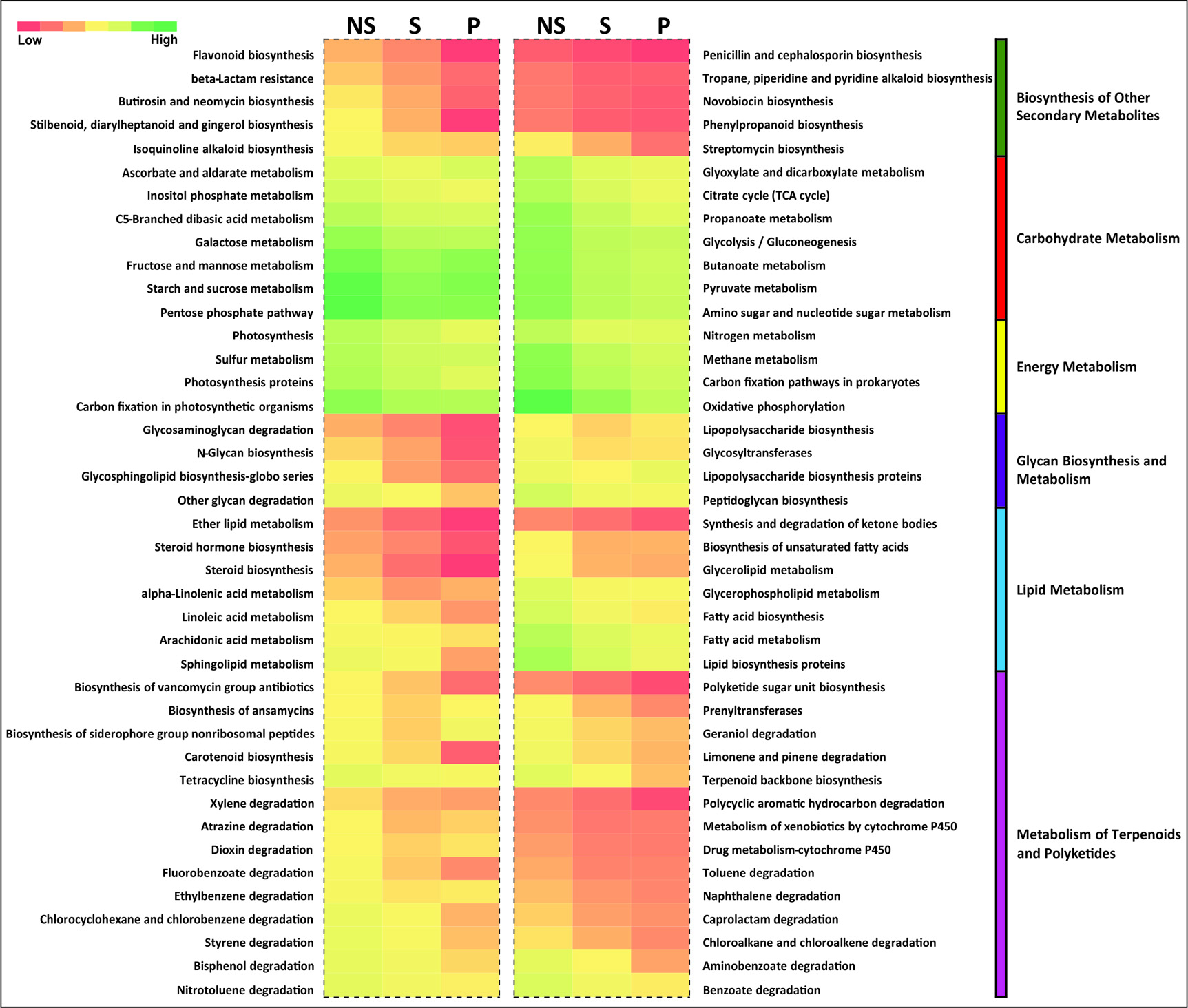
Fig. 4. Functional capacities regarding with metabolism of the root associated soil microbiome. This heatmap showed relative abundance of representative functions which are derived from the PICRUSt tool and greengene 13.8 database.
This culture-independent molecular investigation offers new visions into the structure and composition of native microbial communities Colocasia esculenta associated root zones. Importantly, the species level identification was revealed with the dual approach.
Methods to calculate approximately the microbial diversity yet have advanced to comprehend the population and diversity of microorganisms in varied ecosystems26. The 16S rRNA sequences based characterization of microbial communities has been accepted and adopted as the standard method in microbial ecology and a vast number of up-to-date open-source sequence analysis tools such as “mothur”27, “QIIME”28, or “RDP”29 assist the analysis of the large number of sequences generated by modern, advanced and colossal parallel sequencing methods under the umbrella of Next Generation Sequencing (NGS). However, one of the shortcomings of NGS methodologies is the short read length and that sequencing the complete 16S rRNA gene of all-inclusive communities is still expensive and operationally convoluted. The present study follows the traditional approach of generating clone banks followed by the Sanger sequencing method for studying the microbial ecology of the root compartments of Colocasia esculenta. The method to utilize PCR products generated by a single primer pair rather than using separate primer pairs for every variable 16S rRNA region reduces the impact of different primer pairs on the constitution of the PCR products, which can impinge on species composition and species richness (SR) estimates. The method of analysis involves the formation of OTUs based on the reference database SILVA followed by taxonomic assignments using the EzTaxon-e server. Since the clone libraries generated were sequences of length more than 850 bp, it was preferred to complete the taxonomical classification through BLAST analysis using the EzTaxon-e server. The higher cut-off percentage (97%) and e-value were used to minimize the misidentification of the taxa.
The diversity of microbes inhabiting the numerous niches of the root and rhizosphere differs12. Study of the Arabidopsis rhizosphere and bulk soil using 16S rRNA sequencing did not indicate any major difference in the microbial communities, whereas, only a minor decrease was observed in the OTU richness of the rhizosphere microbial population30. Meta-transcriptomic analysis of the rhizosphere and bulk soil indicated that the soil compartments do not show considerable variation in microbial diversity when only the numbers of microbial taxa were considered. However, in our study, a clear distinction between rhizosphere and bulk soil diversity was observed at the genus level and not the phylum level.
Roots offer distinct microhabitats at the soil-root interface: rhizosphere soil, rhizoplane, and endo-rhizosphere12. However, the effectiveness of major nutrients and other micronutrients can be confined to the bacterial population to the Colocasia plant31. Otherwise, rhizosphere effects on the microbial community could be the possible reason to differentiate metabolic potential in the soil to dissolve organic carbon pool32. In the present study, the results indicate a clear distinction in the microbial community diversity at the phylum level as well. The complete absence of members of the phyla Cyanobacteria, Gemmatemonadetes and Planctomycetes from the rhizosphere and rhizoplane microbial population supports the role of root exudates in significantly influencing and designing the microbiome. Recent studies involving the exploration of the rhizoplane population have suggested a greater difference in their bacterial population in comparison to the bulk soil. Experimental studies corroborate the idea that root exudates play a major role in shaping a specialized community which may also have prominent host-specific microbiome signatures. We detected increased relative abundances of Proteobacteria in the rhizosphere and rhizoplane, compared with the bulk soil. The phyla Bacteroidetes and Proteobacteria are by and large deemed as copiotrophic microorganisms (or r-strategists), which grow within environments of high nutrient accessibility33. The copiotrophs can proliferate rapidly in rhizospheric substrate ‘hotspots’ with elevated concentrations of root exudates. Proteobacteria phylum has been showed to have the highest richness (OTUs for frc and oxc,), followed by Actinobacteria and Firmicutes34. Since Colocasia esculenta is an oxalogenic plant and has been known to release oxalates in the root exudates, the preponderance of Proteobacteria in the rhizosphere and rhizoplane microbial communities indicates the oxalotrophic activity which might be a major functional trait of the communities associated. Nunes da Rocha et al. demonstrated that groups associated with Verrucomicrobia might be essential to the rhizosphere of several plants, such as grasses, leek, and potatoes35. On the other hand, a non- Streptomyces actinomycete, isolated from the rhizosphere of an elephant ear plant (Colocasia esculenta) in Bangkok, Thailand which was identified as a novel Saccharomonospora colocasiae sp. strain S265T 36.
The K-shuff analysis indicates the spatial and structural variability between the communities represented by higher AreaK values. Larger the value of AreaK, more distant are the compared communities and that share fewer members between them. The results also predict the compositional difference between the communities. The K-shuff analysis of the three root compartment libraries supported the previous observations statistically. The results maintained the fact that there is a substantial structural difference in the rhizoplane microbial communities in comparison to the other two libraries (rhizosphere (S) and non-rhizosphere (NS)).
Detection of some of the core species which are present throughout the samples from like NS, S, and P suggests that there may negligence of rhizospheric effect governed by host exudates around the roots and could be the example of invadors demonstration. Since oxalogenic plants could act as good source oxalate exudates and of which enrichment of oxalotrophic bacteria as one of the rhizospheric populations. Parallel objectivity was to identify the microbes from the natural environment which have the putative function of oxalotrophy and can possibly be developed into probiotics that can be utilized in the treatment of kidney stones in near future. Studies have indicated a positive role of the probiotics in the management of kidney stones. In lieu of the objective mentioned, microbes exclusive to the three root compartments were identified. This analysis focused only on the members of the Firmicutes, and can be linked to other studies on human subjects showed that the gut microbiome of humans is rich in Firmicutes and Bacteroidetes37. The acclimatization to obtain nutrients and mechanisms of carbohydrate and energy metabolism from the neighboring environment is of major significance to bacteria for their existence in the corresponding ecological niche. Imputed metagenomic analysis performed in this study suggested considerable insight into such mechanisms.
In congruence with the present study, Arabidopsis thaliana has been found to host distinct microbial communities in the root compartments (i.e., rhizosphere, rhizoplane, and endosphere)30,38,39, Oryza sativa40, or Populus deltoides41. Previous studies have corroborated the fact that plants profile their individual microbiome from bulk soil, which is host-specific42. Clone library approach has its an intrinsic limitation for bulk sequences generation, we anticipate the next-generation sequencing platforms for longer reads but, we revealed the eubacterial soil population for its species-level diversity found around the Colocasia.
The present study attempts to explore the root-associated microbial players in the Colocasia esculenta. It is observed that the rhizoplane, owing to the characteristic root exudates, has a distinctive composition of microbial partners as compared to the rhizosphere and bulk soil communities. Field experiments and an intensive microbial community analysis are required to enhance our knowledge of the Colocasia root-associated bacterial flora.
Additional file:
Supplementary File S1
Supplementary File S2
Supplementary File S3
ACKNOWLEDGMENTS
MVS and SSB acknowledge the Council of Scientific and Industrial Research (CSIR) and University Grant Commission (UGC) respectively for the research fellowships during the Ph.D. program. MVS acknowledges to SERB-National Postdoctoral Fellowship, India (PDF/2018/000147) and, NB acknowledges Dr. DS Kothari Postdoctoral Fellowship of University Grant Commission (UGC), India for providing financial assistance. Authors are thankful to the Department of Biotechnology (DBT), Government of India; National Centre for Microbial Resource (Formerly Microbial Culture Collection) Project (BT/PR10054/NDB/52/94/2007) for financial aids. All authors are thankful to Dr. Srikant Pawar for his guidance and help in sample collection.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
AUTHORS’ CONTRIBUTION
MVS and SSB contributed to the data generation and drafted the manuscript. NB, VC, YSS interpretation supervised and reviewed the manuscript. NB, VC, and PC helped for the preparation of figures, interpretation and draft improvisation. All the authors read and approved the manuscript.
FUNDING
Authors are thankful to the Department of Biotechnology (DBT), Government of India; National Centre for Microbial Resource (Formerly Microbial Culture Collection) Project (BT/PR10054/NDB/52/94/2007) for financial aids. MVS acknowledges to SERB-National Postdoctoral Fellowship (PDF/2018/000147) for Article Processing Charges (APC) of the current manuscript.
ETHICS STATEMENT
This article does not contain any studies with human participants or animals.
AVAILABILITY OF DATA
All datasets generated or analyzed during this study are included in the manuscript. All the 16S rRNA gene sequences generated during this study were deposited in the Genbank-NCBI database under the accession numbers: KF784899 – KF785750.
- Zaitsev GM, Tsitko IV, Rainey FA, Trotsenko YA, Uotila JS, Stackebrandt E, et al. New aerobic ammonium-dependent obligately oxalotrophic bacteria: description of Ammoniphilus oxalaticus gen. nov., sp. nov. and Ammoniphilus oxalivorans gen. nov., sp. nov. Int J Syst Evol Microbiol. 1998; 48: 151–163.
Crossref - Anbazhagan K, Edward Raja C, Selvam GS. Oxalotrophic Paracoccus alcaliphilus isolated from Amorphophallus sp. rhizoplane. World J Microbiol Biotechnol. 2007; 23: 1529–1535.
Crossref - Kost T, Stopnisek N, Agnoli K, Eberl L, Weisskopf L. Oxalotrophy, a widespread trait of plant-associated Burkholderia species, is involved in successful root colonization of lupin and maize by Burkholderia phytofirmans. Front Microbiol. 2014; 4.
Crossref - Franceschi VR, Nakata PA. Calcium oxalate in plants: formation and function. Annu Rev Plant Biol. 2005; 56: 41–71.
Crossref - Abratt VR, Reid SJ. Oxalate-degrading bacteria of the human gut as probiotics in the management of kidney stone disease. Adv Appl Microbiol. 2010. 63–87.
Crossref - Jones DL. Organic acids in the rhizosphere – a critical review. Plant Soil. 1998; 205: 25–44.
Crossref - Garvie LAJ. Decay of cacti and carbon cycling. Naturwissenschaften. 2006; 93: 114–118.
Crossref - Noonan SC, Savage GP. Oxalate contents of food and its effect on humans. Asia Pacific J Clin Nutr. 1999; 8(1): 64-74.
Crossref - Pereira P, Ibanez F, Rosenblueth M, Etcheverry M, Martinez-Romero E. Analysis of the bacterial diversity associated with the roots of maize (Zea mays L.) through culture-dependent and culture-independent methods. ISRN Ecol. 2011.
Crossref - Suryavanshi MV, Bhute SS, Jadhav SD, Bhatia MS, Gune RP, Shouche YS. Hyperoxaluria leads to dysbiosis and drives selective enrichment of oxalate metabolizing bacterial species in recurrent kidney stone endures. Sci Rep. 2016; 6: 1–15.
Crossref - Marathe N, Shetty S, Lanjekar V, Ranade D, Shouche Y. Changes in human gut flora with age: an Indian familial study. BMC Microbiol. 2012; 12: 222.
Crossref - Reinhold-Hurek B, Bunger W, Burbano CS, Sabale M, Hurek T. Roots shaping their microbiome: global hotspots for microbial activity. Annu Rev Phytopathol. 2015; 53: 403–424.
Crossref - Suryavanshi MV, Bhute SS, Bharti N, Pawar K, Shouche YS. Eubacterial diversity and oxalate metabolizing bacterial species (OMBS) reflect oxalate metabolism potential in odontotermes gut. J Pure Appl Microbiol. 2016; 10: 2035–44.
- Ashelford KE, Chuzhanova NA, Fry JC, Jones AJ, Weightman AJ. New screening software shows that most recent large 16S rRNA gene clone libraries contain chimeras. Appl Environ Microbiol. 2006; 72: 5734–5741.
Crossref - Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007; 23: 2947–2948.
Crossref - Ashelford KE, Chuzhanova NA, Fry JC, Jones AJ, Weightman AJ. At least 1 in 20 16S rRNA sequence records currently held in public repositories is estimated to contain substantial anomalies. Appl Environ Microbiol. 2005; 71: 7724–7736.
Crossref - Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010; 7: 335–336.
Crossref - Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010; 26: 2460–2461.
Crossref - Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013; 41: D590–D596.
Crossref - Chun J, Lee J-H, Jung Y, Kim M, Kim S, Kim BK, et al. EzTaxon: a web-based tool for the identification of prokaryotes based on 16S ribosomal RNA gene sequences. Int J Syst Evol Microbiol. 2007; 57: 2259–2261.
Crossref - Langille MGI, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. 2013; 31: 814–821.
Crossref - Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000; 28: 27–30.
Crossref - Parks DH, Beiko RG. Identifying biologically relevant differences between metagenomic communities. Bioinformatics. 2010; 26: 715–721.
Crossref - Swift ML. GraphPad Prism, data analysis, and scientific graphing. J Chem Inf Comput Sci. 1997; 37: 411–412.
Crossref - Jangid K, Kao M-H, Lahamge A, Williams MA, Rathbun SL, Whitman WB. K-shuff: A novel algorithm for characterizing structural and compositional diversity in gene libraries. Plos one. 2016; 11: e0167634.
Crossref - Birtel J, Walser J-C, Pichon S, Burgmann H, Matthews B. Estimating bacterial diversity for ecological studies: methods, metrics, and assumptions. Plos one. 2015; 10: e0125356.
Crossref - Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009; 75: 7537–7541.
Crossref - Kuczynski J, Stombaugh J, Walters WA, Gonzalez A, Caporaso JG, Knight R. Using QIIME to analyze 16S rRNA gene sequences from microbial communities. Curr Protoc Microbiol. 2012; 27: 1E.5.1-1E.5.20.
Crossref - Maidak BL, Cole JR, Parker CT, Garrity GM, Larsen N, Li B, et al. A new version of the RDP (Ribosomal Database Project). Nucleic Acids Res. 1999; 27: 171–173.
Crossref - Lundberg DS, Lebeis SL, Paredes SH, Yourstone S, Gehring J, Malfatti S, et al. Defining the core Arabidopsis thaliana root microbiome. Nature. 2012; 488: 86–90.
Crossref - Laxminarayana K. Response of mycorrhiza, organic sources, secondary and micro nutrients on soil microbial activities and yield performance of Colocasia (Colocasia esculenta L.) in alfisols. Commun Soil Sci Plant Anal. 2016; 47: 775–786.
Crossref - Song M, Cheng Z, Luo C, Jiang L, Zhang D, Yin H, et al. Rhizospheric effects on the microbial community of e-waste-contaminated soils using phospholipid fatty acid and isoprenoid glycerol dialkyl glycerol tetraether analyses. Environ Sci Pollut Res. 2018; 25: 9904–9914.
Crossref - Fierer N, Bradford MA, Jackson RB. Toward an ecological classification of soil bacteria. Ecology. 2007; 88: 1354–1364.
Crossref - Herve V, Junier T, Bindschedler S, Verrecchia E, Junier P. Diversity and ecology of oxalotrophic bacteria. World J Microbiol Biotechnol. 2016; 32: 28.
Crossref - Rocha UN da, Plugge CM, George I, Elsas JD van, Overbeek LS van. The rhizosphere selects for particular groups of acidobacteria and verrucomicrobia. Plos one. 2013; 8: e82443.
Crossref - Wattanasuepsin W, Intra B, Take A, Inahashi Y, Euanorasetr J, Omura S, et al. Saccharomonospora colocasiae sp. nov., an actinomycete isolated from the rhizosphere of Colocasia esculenta. Int J Syst Evol Microbiol. 2017; 67: 4572–4577.
Crossref - Mahowald MA, Rey FE, Seedorf H, Turnbaugh PJ, Fulton RS, Wollam A, et al. Characterizing a model human gut microbiota composed of members of its two dominant bacterial phyla. Proc Natl Acad Sci. 2009; 106: 5859–5864.
Crossref - Bulgarelli D, Rott M, Schlaeppi K, Ver Loren van Themaat E, Ahmadinejad N, Assenza F, et al. Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature. 2012; 488: 91–95.
Crossref - Bodenhausen N, Horton MW, Bergelson J. Bacterial communities associated with the leaves and the roots of Arabidopsis thaliana. Plos one. 2013; 8: e56329.
Crossref - Edwards J, Johnson C, Santos-Medellin C, Lurie E, Podishetty NK, Bhatnagar S, et al. Structure, variation, and assembly of the root-associated microbiomes of rice. Proc Natl Acad Sci. 2015; 112: E911–E920.
Crossref - Gottel NR, Castro HF, Kerley M, Yang Z, Pelletier DA, Podar M, et al. Distinct microbial communities within the endosphere and rhizosphere of Populus deltoides roots across contrasting soil types. Appl Environ Microbiol. 2011; 77: 5934–5944.
Crossref - Ofek-Lalzar M, Sela N, Goldman-Voronov M, Green SJ, Hadar Y, Minz D. Niche and host-associated functional signatures of the root surface microbiome. Nat Commun. 2014; 5: 1–9.
Crossref
© The Author(s) 2020. Open Access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License which permits unrestricted use, sharing, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.