


ISSN: 0973-7510
E-ISSN: 2581-690X
Computational databases and tools in recent times have been proved to provide an essential aid for anticancer studies in the field of oncology. Molecular docking studies facilitate the structural diversity of plant-derived phytomolecules having anticancer properties against receptor proteins involved in cancer signaling pathways. The current study involves the investigation of phytocompounds-agasthisflavone, anacardic acid, zoapatanolide A, a purified product of the plant extract Amarogopinois546 were subjected to docking studies on p38-α MAPK and EGFR Kinase domain. The effectiveness of this study was evaluated by comparing the docking interactions of a standard drug, doxorubicin against the receptor molecules. The docking study is analyzed by compound estimated with lowest binding energy is considered to have the highest affinity towards the active site of the receptor proteins. The isolated plant compound Amarogopinois546 displayed the least binding score with a large number of hydrogen bonds and hydrophobic interactions towards the P38α MAP kinase receptor in comparison with the EGFR kinase domain. This preliminary result can strongly be supported for carrying out experimental evaluation in near future.
Phytomolecules, Docking, Binding Energy, Hydrophobic, MAPK, EGFR
Ayurveda is a sacred precept of rule or commandments which guides us about all aspects of life and it is considered to be the oldest healing science in India. Anacardium occidentale commonly known as Cashew belongs to the family Anacardiaceae considered as a multi-purpose tree since the leaves and the bark of the tree are of therapeutic importance and the fruit is an edible resource of high value. Anacardium occidentale native of Brazil is an all-purpose tree that is widely distributed in India, Brazil, Africa, Mexico, Malaysia, and Turkey. Vitamin B2 and B3, vitamin E, vitamin C, pantothenic acid and magnesium are all abundant in cashews. Zoapatanolide A, Agasthisflavone, and anacardic acid are the biological components identified in substantial quantities in the powdered leaf extract and Cashew nutshell liquid, These phytochemicals were checked for their in-silico anticancer activity.
Molecular Docking is a computational in-silico approach that helps in predicting the preferred orientation of a molecule (ligand) when it is made to interact at the binding pocket of a macromolecule (enzyme/protein)1. This knowledge of orientation prediction may in turn help in predicting the binding affinity or the strength of association between two molecules2. The in-silico molecular docking has become an efficient technique in the field of drug discovery for structure-based medication disclosure3. The most popular and commonly used software for molecular docking purposes is AutoDock (v.4.0). It is freely available open-source software for virtual screening of small molecules to receptors and computational docking4. To obtain preliminary data, docking studies were conducted using the screened phytocompounds as ligands and the target molecules p38-MAPK (p38-mitogen-activated protein kinase) and EGFR (Epidermal growth factor receptor) Kinase domain (3W2S). The target protein, p38 mitogen-activated protein kinase belongs to serine/threonine protein kinases. The human p38 MAP kinase is classified into four isoforms: p38α, p38β, p38γ and p38δ, with p38α being the most well-studied of the four. Phosphorylation of nuclear and cytoplasmic targets is carried out by the p38 MAP kinase5. p38 MAP kinase is a signaling pathway that regulates a complex network of proteins involved in cellular activity, cell senescence, cell cycle arrest, cell differentiation, tumor suppression, apoptosis and cytokine synthesis6,7.
Protein preparation
The structure of proteins, 4FA2 (mitogen-activated protein kinase) and 3W2S (epidermal growth factor receptor) were obtained from Protein Data Bank (PDB) 8 and proteins devoid of the ligand is represented in Fig. 1. The protein structures were validated through the Ramachandran plot depicted in Fig. 2. Only structures which had at least ~96% of residues in favored regions and ~2% of residues in allowed regions were fit for molecular interaction studies. Ramachandran plot prediction for the structure of p38α MAP kinase protein showed 97.9% residues in favored regions and for EGFR Kinase domain protein 97.7% residues in favored regions9.
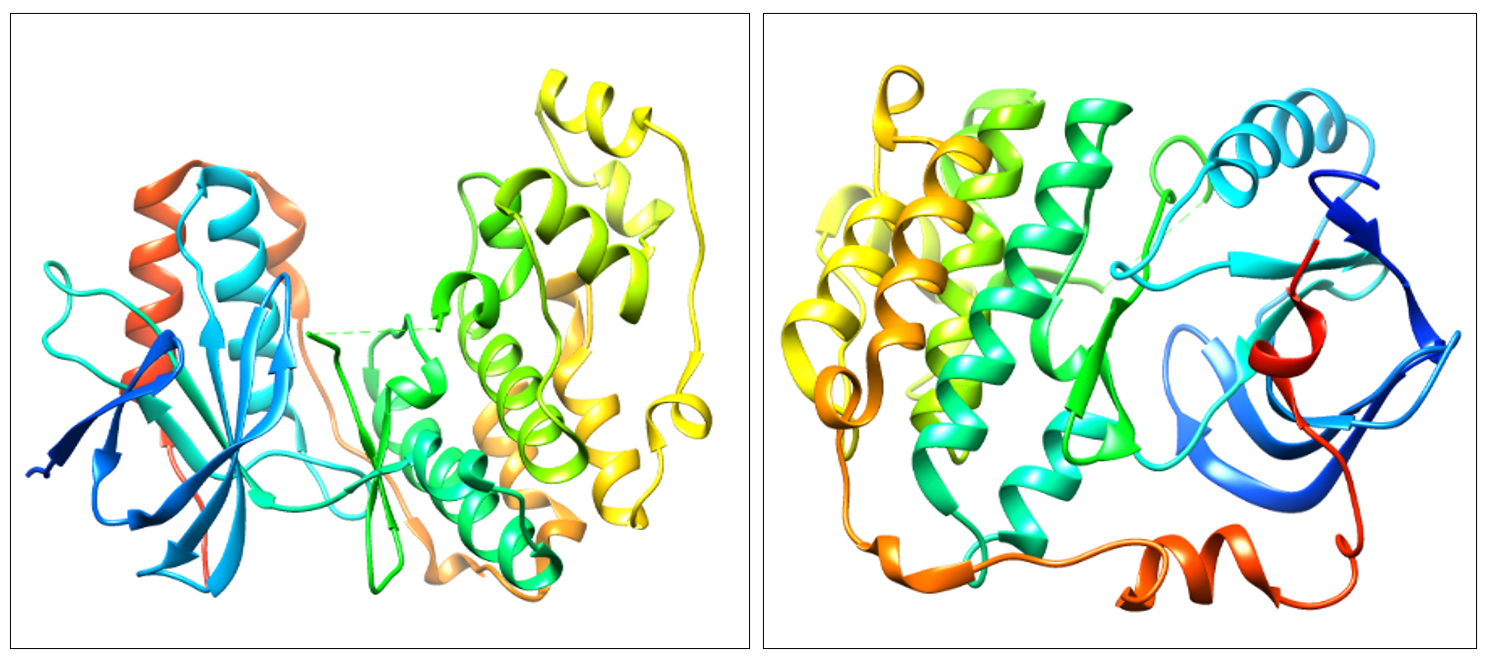
Fig. 1. 3D structure of P38α MAP kinase (4FA2) and EGFR Kinase domain (3W2S).
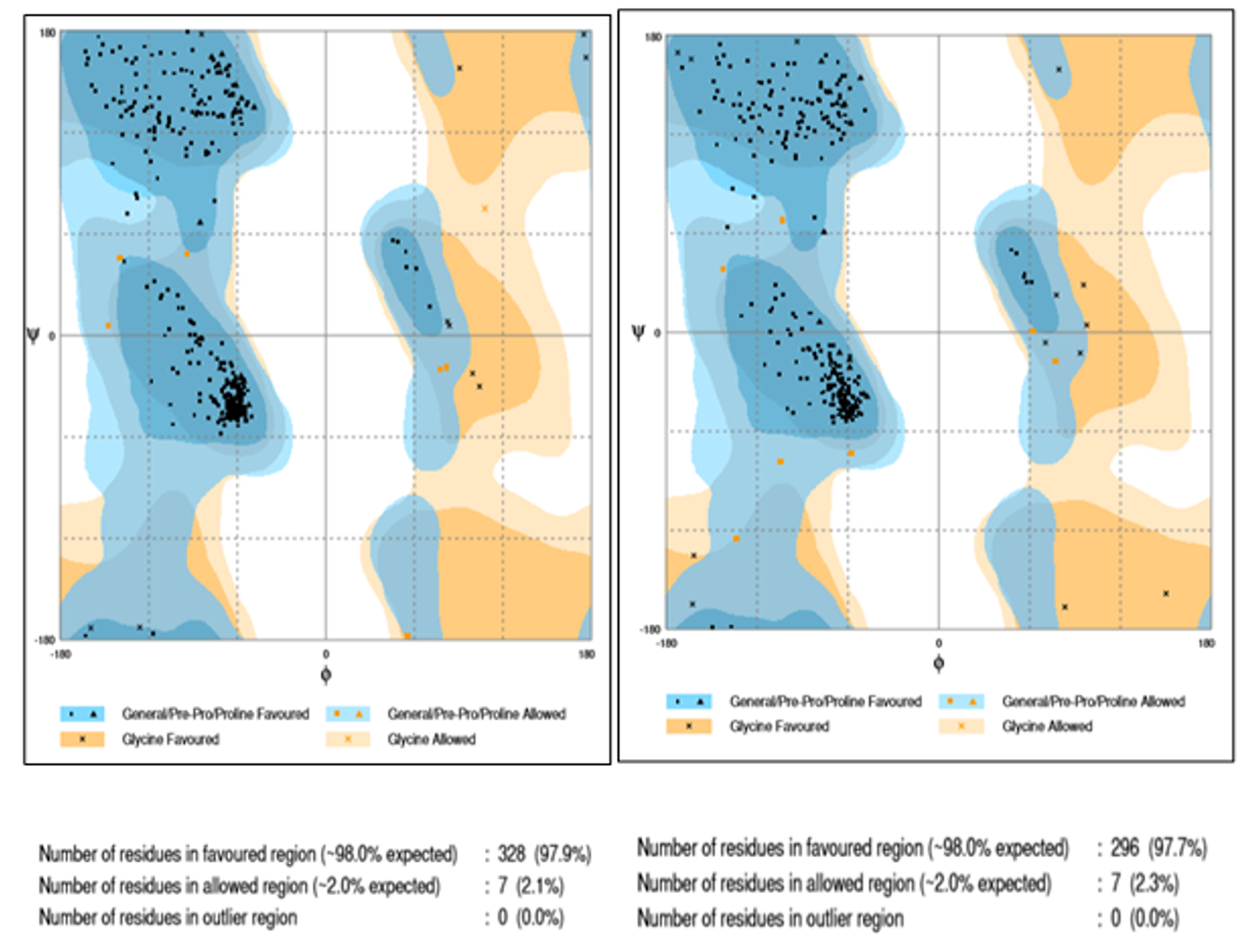
Fig. 2. Ramachandran plot prediction for structure 4FA2 and 3W2S.
Ligand Preparation
The structure of agasthisflavone, anacardic acid, zoapatanolide A and standard drug doxorubicin were obtained from Pubchem. The obtained SDF format of the above-mentioned ligands was converted to .pdb format using Open Babel (2.4.0) by adding hydrogens and generating 3D coordinates10. The purified product of the plant extract Amarogopinois546 was drawn using Chemsketch and saved in .mdl format which was further converted into .pdb format11. The geometry of all these PDB structures was cleaned and refined using Argus Lab (4.0.1)12. The validated structure which obeyed structure-activity relationships (SAR) was selected and the structures of agasthisflavone, anacardic acid, zoapatanolide A, doxorubicin and the Amarogopinois546 are represented in Fig. 3.
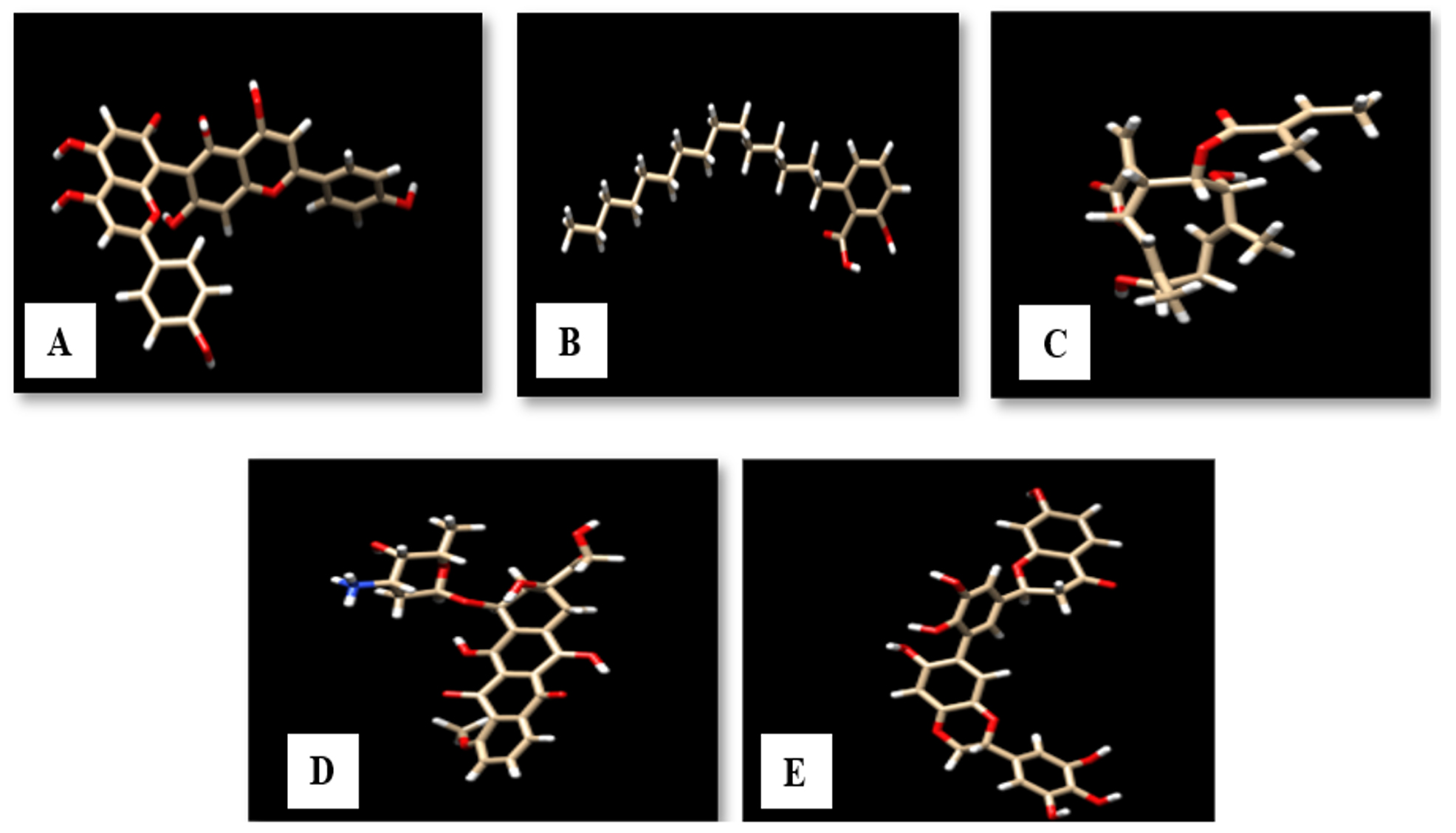
Fig. 3. 3D structure of A: Agasthisflavone, B: Anacardic acid, C: Zoapatanolide A, D: Doxorubicin and E: Amarogopinois546.
The binding residues for the proteins P38α MAP kinase (4FA 2) and EGFR Kinase domain (3W2S) were predicted using the GalaxySite tool of GalaxyWEB server13. Each protein showed the involvement of more than 6 residues in the formation of the active site (Table 1).
Table (1):
The details of amino acid residues of both 4FA2 and 3W2S.
No. |
Protein PDB ID |
Binding site residues |
|---|---|---|
1 |
4FA2 |
30V, 38V, 53K, 71E, 75L, 84I, 104L, 106T, 109M, 110G, 111A, 167L, 168D, 169F |
2 |
3W2S |
718L, 726V, 743A, 745K, 795F, 796G, 797C, 800D, 801Y, 841R, 842N, 844L, 854T, 997F |
Molecular Docking
The predicted protein structures docked using Autodock suite (v.4.0)14. Autodock is a set of automated docking tools, which predicts the interactions between a small molecule and its receptor proteins, which proposes several binding modes of a docked complex15,16. It employs the Lamarckian genetic algorithm, with Gasteiger performing the charge computations. All of the chosen ligands were docked using a Genetic algorithm, in which the ligands were subjected to conformational modifications to find the lowest energy conformation, considered as the most stable structure16. The binding modes with the lowest energies were screened and grouped. The chosen compounds were docked against the target proteins to identify and evaluate the binding affinity of the docked complexes18,19. The molecular docking complexes were further visualized using the UCSF Chimera visualization tool to manually pick the best feasible docking pose20.
Table (2):
Residues of the target molecule p38α MAP kinase interacting with the three ligands: Zoapatanolide A, Agasthisflavone and Anacardic acid, a standard drug Doxorubicin and Amarogopinois546.
| Name of the Biomarker | Ligands | H-bond interaction | Amino acid residues of targe | |
|---|---|---|---|---|
| No. of bonds | Amino acid Residues | (Hydrophobic interactions) | ||
|
p38α MAP kinase
|
Agasthisflavone
|
4 | Ala 157 Leu 171 Gly 31 Leu 156 |
Ala 157, Leu 156, Asn 155, Pro 153, Asp 112, Ser 154, Gly 170, Leu 171, Leu 104, Lys 53, Val 52, Val 38, Gly 31, Val 30, Ser 32 |
|
Anacardic acid
|
1 | UNL | Leu 74, Leu 75, Ile 84, Ile 166, Leu 167, Asp 168, Phe 169, Glu 71, Thr 106, Ala157 | |
|
Zoapatanolide A
|
2 | UNL Ser 154 |
Ser 154, Lys 152, Asn 155, Leu 171 | |
| Doxorubicin (standard drug) |
4 | UNL UNL Lys 53 Gly 170 |
Phe 169, Gly 170, Lys 53, Leu 171, Gly 33, Ser 32, Ser 154, Gly 31, Val 163, Lys 162 | |
| Amarogopinois546 | 6 | UNL UNL Met 109 Gly 110 Leu 156 Lys 165 |
Thr 106, His 107, Leu 108, Ile 84, Lys 165, Gly 110, Leu 167, Val 158, Ala 157, Leu 156, Ala 111, Asn 155, Ser 154, Pro 153, Asp 112, Leu 113, Gly 170, Leu 171, Gly 33, Ser 32 | |
Table (3):
Residues of the target molecule EGFR Kinase domain interacting with Zoapatanolide A, Agasthisflavone, Anacardic acid, a standard drug Doxorubicin and Amarogopinois546.
| Name of the Biomarker | Ligands | H-bond interaction | Amino acid residues of target proteins (Hydrophobic interactions) |
|
|---|---|---|---|---|
| No. of bonds | Amino acid Residues | |||
|
EGFR Kinase domain
|
Agasthisflavone
|
4 | UNL UNL Ala 722 Met 793 |
Ala 157, Leu 156, Asn 155, Pro 153, Asp 112, Ser 154, Gly 170, Leu171, Leu 104, Lys 53, Val 52, Val 38, Gly 31, Val 30, Ser 32 |
|
Anacardic acid
|
3 | Ala 722 Phe 723 Gly 724 |
Leu 74, Leu 75, Ile 84, Ile 166, Leu 167, Asp 168, Phe 169, Glu 71, Thr 106, Ala157 | |
|
Zoapatanolide A
|
2 | Ala 722 Gly 724 |
Ser 154, Lys 152, Asn 155, Leu 171 | |
| Doxorubicin (standard drug) |
4 | UNL UNL Ala 722 Phe 723 |
Phe 169, Gly 170, Lys 53, Leu 171, Gly 33, Ser 32, Ser 154, Gly 31, Val 163, Lys 162 | |
|
Amarogopinois546 |
5 | UNL UNL LYS 745 MET 793 ARG 803 |
Leu 718,Gly 719, Ser 720, Val 726, Phe 997, Met 793, Gly 796, Asp 800, Arg 803, Leu 1001, Leu 844, Lys 745, Asp 855 | |
The molecular interaction studies performed through molecular docking using Autodock (4.2.6) of agathisflavone, anacardic acid, zoapatanolide A, doxorubicin and the Amarogopinois546 against the P38α MAP kinase (4FA2) and EGFR Kinase domain (3W2S) exhibited binding affinity and molecular interactions with proteins (Table 2 and 3). Whereas, binding affinity towards P38α MAP kinase (4FA2) showed significant binding towards all the ligand molecules. All the interactions were visualized using UCSF Chimera21 and the depicted pictures were shown in Figs. 4 and 5.
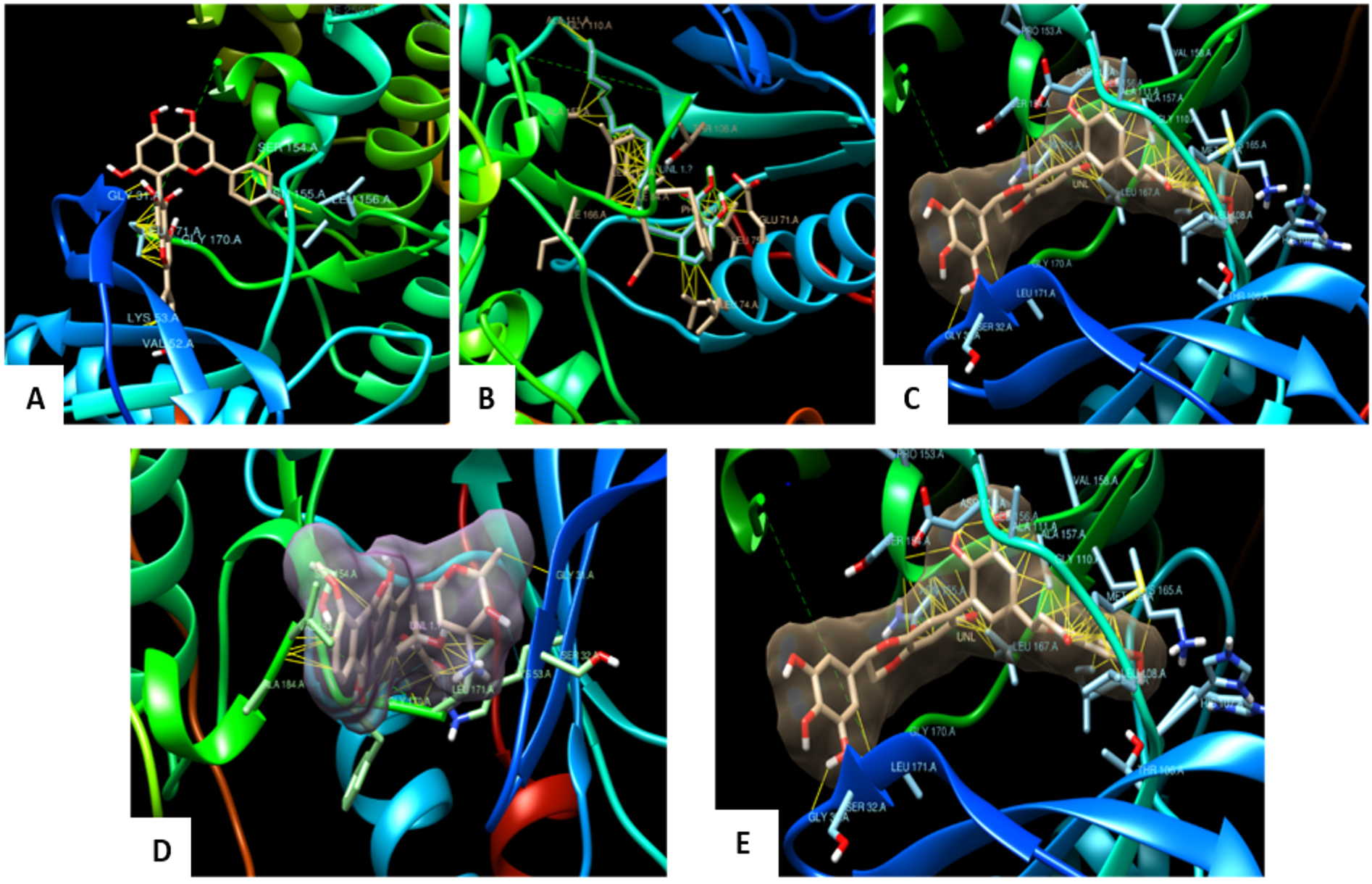
Fig. 4. Docked pose of the ligand shown with p38α MAP kinase protein and Hydrophobic interactions displayed within the solid surface of the docked complex between the binding residues of the p38α MAP kinase receptor and the small molecule A: Agasthisflavone, B: Anacardic acid, C: Zoapatanolide A, D: Doxorubicin and E: Amarogopinois546.
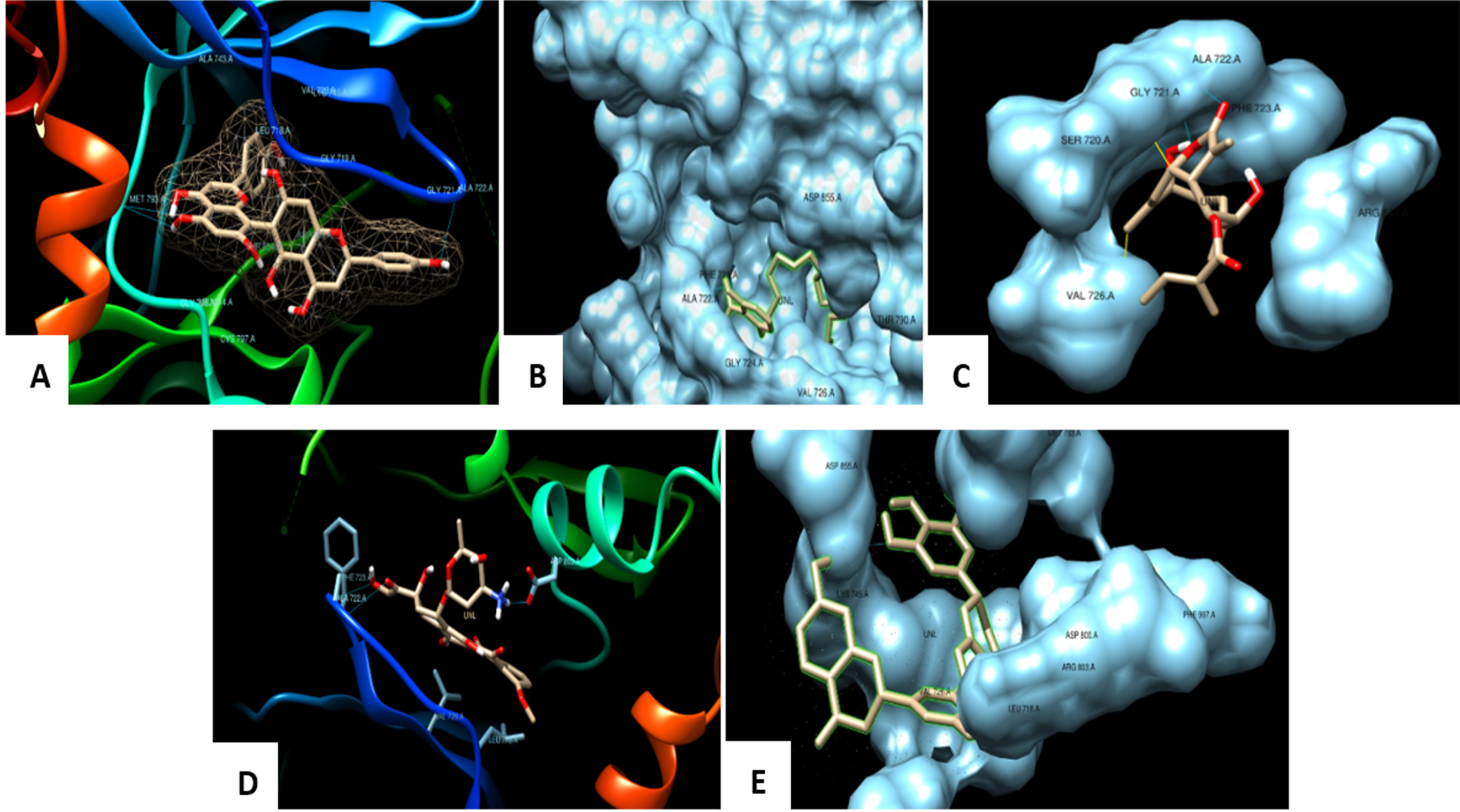
Fig. 5. Docked pose of the ligand shown with EGFR Kinase protein and the binding pocket of the EGFR protein fits the ligand by interactions and bonds A: Agasthisflavone, B: Anacardic acid, C: Zoapatanolide A, D: Doxorubicin and E: Amarogopinois546
Ligand interactions with 4FA2 and 3W2S
All the ligands selected have shown a very high binding affinity and low binding energy towards 4FA2 protein when compared to 3W2S protein. However, the screened phytomolecule Amarogopinois546 has shown a greater number of hydrogen bonds and hydrophobic interactions with both 4FA2 and 3W2S when compared to other ligands and standard drug doxorubicin. Thus, by making it a novel compound.
From the above interaction studies results obtained, the main isolated phytochemical Amarogopinois546 exhibited high binding affinity and lowest binding energy with the formation of a greater number of bonded (hydrogen bonds) and non-bonded interactions (hydrophobic interactions) towards the P38α MAP kinase receptor (4FA2) which infers that the isolated phytochemical Amarogopinois546 has interacted very well with both the proteins when compared to the standard drug Doxorubicin (Inhibitor). Researchers have discovered that p38 MAP kinase plays a significant impact in cancer20.
The in silico studies were designed by selecting screened phytocompounds as ligands and target proteins as p38-α MAPK (p38-α mitogen-activated protein kinase) and EGFR Kinase domain (3W2S). The selected ligands were compared with the isolated compound Amarogopinois546 and standard drug doxorubicin. The objective concluded that Amarogopinois546 showed a high binding affinity towards the P38α MAP kinase receptor (4FA2) when compared to the EGFR Kinase domain (3W2S). Thereby concluding all the predefined objectives of this investigation with the following future aspects.
ACKNOWLEDGMENTS
Amar Shankar acknowledges the support and infrastructure provided by School of Engineering and Technology, JAIN (Deemed-to-be University). KSP gratefully thank the Director, Amrita Vishwa Vidyapeetham, Mysuru campus, Mysuru for infrastructural facilities. CS and SKP acknowledge the support and infrastructure provided by the JSS Academy of Higher Education and Research (JSSAHER), Mysuru, India.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
AUTHORS’ CONTRIBUTION
AS, GSM, SPK, SP, ASJ, SSP, CS, and CS conceptualized and gathered the data with regard to this work. AS, CS and SP gave the necessary inputs towards the designing of the manuscript.
FUNDING
None.
ETHICS STATEMENT
Not applicable.
AVAILABILITY OF DATA
All datasets generated or analyzed during this study are included in the manuscript.
- Azam SS, Abbasi SW. Molecular docking studies for the identification of novel melatoninergic inhibitors for acetylserotonin-O-methyltransferase using different docking routines. Theor Biol Med Model. 2013;10:63.
Crossref - Umesh HR, Ramesh KV, Devaraju KS. Molecular docking studies of phytochemicals against trehalose-6-phosphate phosphatases of pathogenic microbes. Beni Suef Univ J Basic Appl Sci. 2020;9:5.
Crossref - Acharya R, Chacko S, Bose P, Lapenna A, Pattanayak SP. Structure Based Multitargeted Molecular Docking Analysis of Selected Furanocoumarins against Breast Cancer. Sci Rep. 2019;9:15743.
Crossref - Forli S, Huey R, Pique ME, Sanner MF, Goodsell DS, Olson AJ. Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat Protoc. 2016;11(5):905-919.
Crossref - Sebolt-Leopold JS. Development of anticancer drugs targeting the MAP kinase pathway. Oncogene. 2000;19(56):6594-6599.
Crossref - Olson MO. Sensing cellular stress: another new function for the nucleolus?. Science Signaling. 2004;224:10.
Crossref - Pearson BJ, Alvarado AS. Regeneration, stem cells, and the evolution of tumor suppression. Cold Spring Harb Symp Quant Biol. 2008;73:565-572.
Crossref - Berman HM, Westbrook J, Feng Z, et al. The Protein Data Bank. Nucleic Acids Res. 2000;28(1):235-242.
Crossref - Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera–a visualization system for exploratory research and analysis. J Computat Chem. 2004;25(13):1605-1612.
Crossref - Wang W, Xia M, Chen J, et al. Data set for phylogenetic tree and RAMPAGE Ramachandran plot analysis of SODs in Gossypium raimondii and G. arboretum. Data in Brief. 2016;9:345-348.
Crossref - Ko J, Park H, Heo L, Seok C. GalaxyWEB server for protein structure prediction and refinement. Nucleic Acids Res. 2012;40(W1):W294-W297.
Crossref - O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open Babel: An open chemical toolbox. J Cheminform. 2011;3:33.
Crossref - Naz A, Bano K, Bano F, Ghafoor NA, Akhtar N. Conformational analysis (geometry optimization) of nucleosidic antitumor antibiotic showdomycin by Arguslab 4 software. Pak J Pharm Sci. 2009;22(1):78-82.
- Dallakyan S, Olson AJ. Small-molecule library screening by docking with PyRx. Methods Mol Biol. 2015;1263:243-50.
Crossref - Prasad A, Shruthi G, Sushma P, et al. Helicobacter pylori infection: a bioinformatic approach. International Journal of Pharmaceutical Sciences and Research. 2020;11(11):1000-1015.
- Chaudhari R, Li Z. PyMine: a PyMOL plugin to integrate and visualize data for drug discovery. BMC Res Notes. 2015;8:517.
Crossref - Jain AS, Sushma P, Dharmashekar C, et al. In silico evaluation of flavonoids as effective antiviral agents on the spike glycoprotein of SARS-CoV-2. Saudi J Biol Sci. 2021;28(1):1040-1051.
Crossref - Dharmashekar C, Pradeep S, Jain AS, et al. Virtual screening of potential phyto-candidates as therapeutic leads against SARS-CoV-2 infection. Environmental Challenges. 2021;4:100136.
Crossref - Prasad SK, Pradeep S, Shimavallu C, et al. Evaluation of Annona muricata Acetogenins as Potential Anti-SARS-CoV-2 Agents Through Computational Approaches. Front Chem. 2021;8:624716.
Crossref - Bursulaya BD, Totrov M, Abagyan R, Brooks CL III. Comparative study of several algorithms for flexible ligand docking. J Comput Aided Mol Des. 2003;17(11):755-763.
Crossref - Avinash KO, Sushma P, Chandan S, et al. In Silico Screened Flavanoids of Glycyrrhiza glabra Inhibit Cpla2 And Spla2 In Lps Stimulated Macrophages. Bull Env Pharmacol Life Sciences. 2021;10(4):14-24.
© The Author(s) 2021. Open Access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License which permits unrestricted use, sharing, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.