


ISSN: 0973-7510
E-ISSN: 2581-690X
Development of a suitable recombinant peptide vaccine against pathogens requires designing of effective immunogenic polypeptide taking various aspects and complexity of immune-response into consideration. Implementing SARS-CoV-2 spike glycoprotein (S-protein) and RNA-dependent RNA polymerase (RdRp) as model targets, in this study, we outline and assess a strategy for in silico recombinant vaccine designing. After mapping the linear B-cell epitopes and MHC1-binding T-cell epitopes six epitopes were sorted from each of the proteins on the basis of extent of residue-conservancy among three types of coronaviruses namely SARS-CoV-2, SARS-CoV and MERS-CoV. Each of the selected epitopes were profiled for their pro-inflammatory potential through molecular docking analysis with surface bound Toll-like receptors, namely TLR2, TLR4 and TLR5. Based on a custom scoring function, the epitopes were ranked for highest and least pro-inflammatory potential. Segments of Spike and RdRp harboring such epitopes were combined using linkers to design immunogenic recombinant polypeptide. Antigenicity and allergenicity of each of the combination was scored; and the best fitting one was docked against TLR2, TLR4 and TLR5 for assessing pro-inflammatory potential. Codon optimization and in silico cloning in expression vector indicated that the designed peptide can be satisfactorily expressed in bacteria, reinforcing the viability of the strategy in identification and designing of potential immunogens.
Spike glycoprotein, RNA-dependent RNA polymerase, Epitopes, Toll-like receptor, combinatorial multiepitope vaccine
COVID-19 pandemic caused by (SARS-CoV-2) started in 2019 December from Wuhan, China has created an unprecedented health-related crisis.1 Currently accounting for 209 million positive cases and 4.4 million deaths worldwide, the vaccination drive is going on successfully to ameliorate the rate of infection. SARS-CoV-2 is a member of β-coronavirus of the Coronaviridae family with genome consist of large positive-sense single-stranded RNA (28.9 kb). Its genome remains encapsulated in a lipid bilayer with Spike glycoproteins adorning the surface like a crown or corona and giving its characteristic name.2 Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) shows almost 79% genomic similarity with SARS-CoV and MERS-CoV variants responsible for two Coronavirus pandemics in 2002 and 2012.
With the onset of COVID-19 pandemic, scientists were able to understand the immunogenic response of SARS-CoV-2 infection and suitable efforts were made to design an effective vaccine.3 About 287 different projects on vaccine development were running in several countries with most of them in the pre-clinical stage. About twelve of them finished human trials and are currently available in the market. The spike glycoprotein of SARS-CoV-2 is one of the attractive vaccine target for being the most antigenic viral protein and experimentally shown to be the strong inducer for CD4+ T cells.3,4 A study reported the potent binding of SARS-CoV S murine polyclonal antibody and it inhibits SARS-CoV-2 S protein in mediating entry into cells.5 Many in-silico immunoinformatics based studies were performed to design multiepitope vaccines. However, the previous strategies did not address the issue of extreme immunogenic response without adding adjuvants.6 Here, we are proposing an approach to create a combinatorial hybrid multiepitope vaccine having B and T-cell epitope peptides from surface spike glycoprotein and endogenous RNA dependent RNA polymerase (RdRp) protein that may show a moderate to high binding affinity with surface TLRs ultimately contribute a mild to good immunogenic response without affecting efficacy. In the current context of new evolving genomic variant of SARS-CoV-2 with mutations in Spike glycoprotein around the world the commercial vaccines efficacies has been affected.7 Therefore we adopted a modified approach to induct epitopes from highly conserved endogenous proteins to develop an effective vaccine candidates against evolving viruses.
Sequence Retrieval and Multi-sequence alignment
Sequences were retrieved from the UniProtKB database. The UniProtKB ID for RNA-dependent RNA Polymerase (RdRp) of SARS-CoV-2, SARS-CoV and MERS-CoV are P0DTD1, P0C6X7 and K9N7C7 respectively. The length of the part of the sequences taken are 932, 932 and 933 amino acid residues for the three coronaviruses respectively. In case of SARS-CoV-2, only chain A was extracted.
The UniProtKB ID for the spike protein are P0DTC2-1 (start position: 13, end position: 1273), P59594 (start position: 14, end position: 1255) and K9N5Q8 (start position: 18, end position: 1353) of SARS-CoV-2, SARS-CoV, MERS-CoV respectively.
The protein models of SARS-CoV-2 for S protein and RdRp are extracted from the RCSB Protein Data Bank having the PDB ID: 7DDD and PDB ID: 6YYT respectively.
The protein sequences thereby obtained was aligned by using Clustal Omega32 server. The alignment file obtained for the two clusters of protein was downloaded in ALN file format. The ALN file was uploaded to ESPript3 server whereby it was again aligned and the solvent accessibility and the secondary structure was determined by uploading the respective protein structures.
The protein structures of TLR2 (PDB ID: 3A79), TLR4 (PDB ID: 3VQ2) and TLR5 (PDB ID: 3J0A) were retrieved from the RCSB Protein Data Bank.
Mapping of T-cell epitopes
The Cytotoxic T Lymphocytes (CTL) epitopes were predicted by using the IEDB server and the tool used was Tepitool. The epitopes were predicted only for the haplotype HLA-A* 02:01. The predicted epitopes were again screened on the basis of their immunogenicity again using the IEDB server tool MHC I Immunogenicity. From there the epitopes having the highest immunogenicity were considered for the subsequent procedures.
Mapping of B-cell epitopes
The B-cell epitopes were predicted by using the IEDB server and its tool BepiPred 2.0. the epitopes predicted were screened on the basis of length of the epitope and solvent accessibility according to the data rendered by ESPript3.
Molecular Docking with TLRs TLR2, TLR4, TLR5
The shortlisted epitopes, both CTL and B-cell epitopes, were docked with cell surface TLRs TLR2, TLR4 and TLR5 using the MDockPep server. The points of interaction of the docking models were noted. Among all the docked models, the top three models of each of spike protein and RdRp having the lowest binding energy were selected. The epitopes with which these interactions were found was selected for the vaccine construction.
Vaccine construction
The part of the protein sequences bearing the most number of the selected epitopes (making most favourable interactions with the cell surface TLRs) were selected. The two parts of protein sequences one coming from RdRp and the other from spike protein were linked using GPGPG linker.
The constructed sequence was checked for antigenicity by Vaxijen 2.0, ANTIGENpro. Next, the sequence was checked for allergenicity by AlergenFP and AllerTop and it also passed as a non-allergen.
In silico cloning
The protein sequence was optimized to its codon sequence using BackTransSeq and optimized for humans as host. The restriction sites of restriction enzymes NdeI and BamHI were added to the N and C terminal of the sequences.
The sequence thus constructed was cloned into pET-16b expression vector in silico and expressed to produce a protein of 618 amino acids.
Secondary structure prediction
The secondary structure of the recombinant antigen was validated using PSIPred.
Molecular docking with surface TLRs
The model of the recombinant antigen was modelled by Swiss-Model and docked with TLRs TLR2, TLR4 and TLR5.
Immunoinformatics approach to design a multi-epitope vaccine
The conventional techniques of developing vaccines using attenuated pathogenic organism or selecting a big antigenic proteins, causing unnecessary intense immunogenic signals with high antigenic load sometimes turns out to be fatal for older recipients.8,9 Current improvement in computational biology and development of several online platforms in immunomics pave an easier and faster in-silico approach of reverse vaccinology.10 Here, we are proposing a method of choosing immunogenic B- and T-cell peptide epitopes from two different proteins of same pathogenic organism in order to elicit both innate and humoral immunity with low antigenic load thus avoiding the increased immune response. Previous studies on using both exogenous and endogenous peptides in vaccine development extend the immunogenic response due to different pathways adopted for the processing of antigens.11-13 First the B-cells and T-Cells epitope for a surface protein and an endogenous protein were predicted using online epitope prediction platforms. The protein sequences of suitable candidates from related species were aligned to analyze the phylogenetic nature of predicted immunogenic epitopes. A mixed population of 6 epitopes each from two antigenic proteins was shortlisted to perform their docking studies against surface Toll-like receptors (TLRs). Among 36 docking studies, three peptides showing high affinity for surface TLRs will be chosen to design a recombinant antigen as a multi-epitope vaccine. Further modeling studies of this recombinant antigen and analyzing their molecular docking against TLRs will predict the success of this vaccine. The overall idea used in this approach is to design a molecule that has (a) both CTL and HTL epitopes from surface as well as endogenous protein. (b) combination of two protein epitopes can make it moderately antigenic with no allergenicity (c) moderate to high binding affinity with TLRs to regulate the immunogenic response. The flow chart of important steps involved in this approach has been shown in Fig. 1.
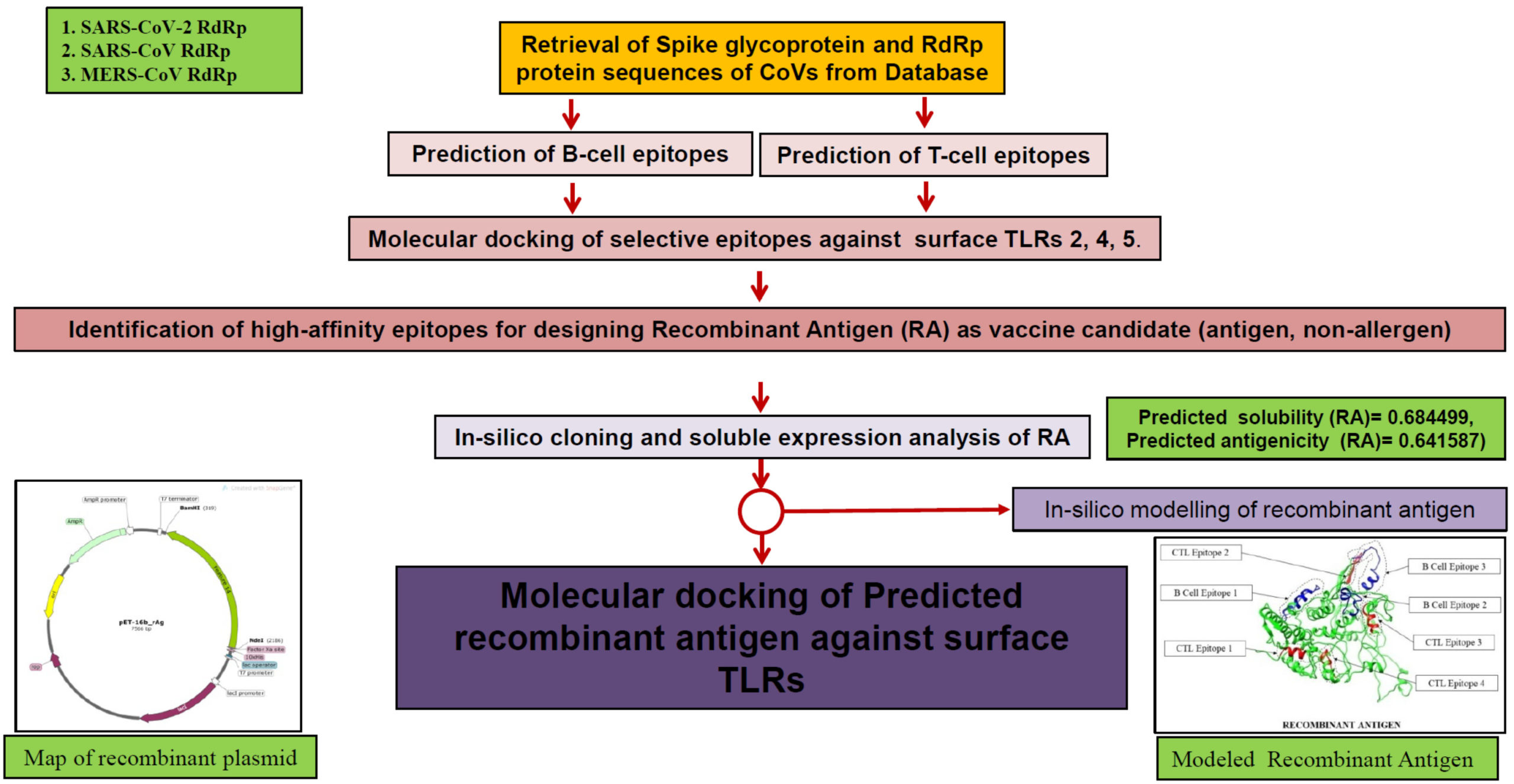
Fig. 1. Flowchart of the approach used in this study. The method adopted here involves utilization of surface and endogenous protein candidates for balanced immunogenic response. The approach is divided in several steps and parameters for screening epitopes having moderate to good binding affinity values while docking with surface expressed Toll-like receptors (TLRs).
Mapping of T-cell and B-cell epitopes for Spike and RNA dependent RNA polymerase (RdRp) proteins
In order to design an effective vaccine, both Cytotoxic T-cell Lymphocytes (CTL) and Helper T-cell Lymphocyte (HTL) epitopes may have a role in inducing natural immunity against the infection which should last for a longer period of time by adaptive immunity. These epitopes are responsible to elicit CD4+ helper response which further leads to development of CD8+ memory T-cells along with activation of B-cells for antibody production.14,15 The protein sequence of spike glycoprotein and RNA dependent RNA polymerase (RdRp) of SARS-CoV-2 were retrieved from open access forum and allowed for prediction of T-cell epitope using suitable software. Additionally, the respective protein sequences from SARS-CoV-2 were also applied for phylogenetic analysis with other evolutionary related viruses like SARS-CoV and MERS-CoV. Matching epitopes with sequence alignment data allowed to identify a mixed population of peptide epitopes having high and low conservation among the variants. The HLA A* 02:01 haplotype CTL epitopes predicted by Tepitool were shortlisted in Supplementary Table1 based on their percentile rank.16 The criteria that epitopes with lower percentile rank have high binding affinity, was considered according to the algorithm used by the server for epitope prediction. Top 6 candidates from each protein were chosen with lower percentile ranks represented in Sup. Table 2. The identified peptide epitopes were further analyzed for their position in protein structures of Spike glycoprotein (PDB: 7DDD) and RdRp (PDB: 7BV1) for solvent accessibility. The protein structures representing the position of identified epitopes and their sequence alignment analysis is shown in Fig. 2. Finally, on the basis of highest score, three such epitopes were chosen for future molecular docking studies with surface TLRs namely TLR2, TLR4 and TLR5. The epitope sequences details of chosen candidate for docking studies is given in Table 1.
Table (1):
Shortlisted epitopes for multiepitope vaccine designing.
No. |
RNA dependent RNA polymerase |
No. |
Spike protein |
|---|---|---|---|
1 |
4456NLIDSYFVV4464(4456R4464) |
1 |
269YLQPRTFLL277(269S277) |
2 |
4515TMADLVYAL4523(4515R4523) |
2 |
417KIADYNYKL425(417S425) |
3 |
4726FVDGVPFVV4734(4726R4734) |
3 |
976VLNDILSRL984(976S984) |
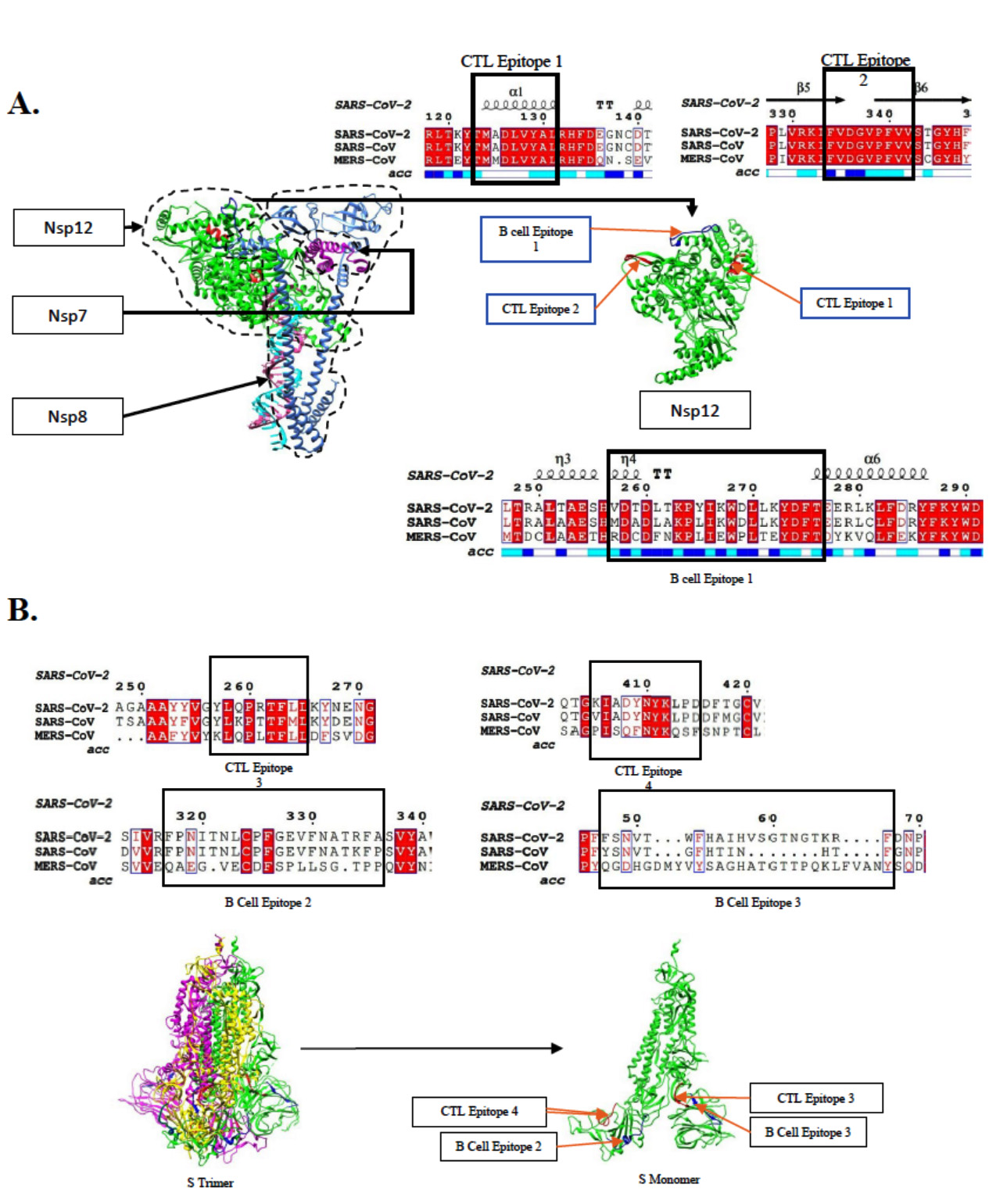
Fig. 2. Phylogenetic analysis of selective epitopes by multiple sequence alignment of epitopes rendered on Espript3.0. Sequence conservation analysis and position of B-cell (blue) and T-cell (red) epitope identified for (A). RNA dependent RNA polymerase enzyme (PDB: 7BV1) (B) Spike glycoprotein (PDB: 7DDD).
The shortlisted epitopes were mapped with protein sequence alignment data which indicates that all the three CTL epitopes mapped on RdRp were quite conserved. While in case of spike glycoprotein, the CTL epitopes were found to be moderately conserved. The CTL epitopes mapped were not found to contain regions where the virus is reported to have undergone mutations. Hence, the epitopes may serve very well as vaccine candidates for the mutant viruses as well.
Th-cell epitopes were accordingly mapped with Tepitools for parameters adjusted to select 21 most predominant MHCII allele. 21 quality epitopes could be mapped in RdRp while 28 such epitopes were predicted for Spike. The peptides from both the proteins were determined to be moderately conserved (Table 2).
Table (2):
Shortlisted B-cell epitopes for multiepitope vaccine designing.
No. |
RNA dependent RNA polymerase |
No. |
Spike protein |
|---|---|---|---|
1 |
154DDYFNKKDWYDFVENP169 |
1 |
59FSNVTWFHAIHVSGTNGTKR78 |
2 |
211DLNGNWYDFGDFIQTTPGSG230 |
2 |
304KSFTVEKGIYQTSNFRVQP322 |
3 |
257VDTDLTKPYIKWDLLKYDFT276 |
3 |
329FPNITNLCPFGEVFNATRFA348 |
4 |
405VAFQTVKPGNFNKDFYDFAV424 |
4 |
672ASYQTQTNSPRRARSVASQ690 |
5 |
444QDGNAAISDYDYYRNLPMCDI465 |
5 |
694AYTMSLGAENSVAYSNN710 |
6 |
497NLDKSAGFPFNKWGKARLYY516 |
6 |
1104VTQRNFYEPQIITTDNTF1121 |
Similarly, B-cell epitopes were predicted using the IEDB server tool BepiPred 2.0,17 with 19-20 residues in length and mostly solvent accessible as indicated by the multiple sequence alignment analysis of SARS-CoV-2, SARS-CoV and MERS-CoV proteins. It is of prime importance for the B-cell epitopes to be solvent accessible and lie exposed on the Spike glycoprotein and RdRp in order to form complex with neutralizing antibodies. The B-cell epitopes considered here are of linear continuous type. Six B-cell epitopes shown in Table 2 were chosen each from spike glycoprotein and RdRp based on their score where higher scores are indicative of their probability of being an epitope. The protein sequence of twelve shortlisted B-cell epitopes are shown in Table 2.
Molecular docking of predicted epitopes with surface TLRs
Surface TLRs (TLR2, TLR4 & TLR5) were chosen to perform the initial molecular docking studies. These particular TLRs being on the surface of the DC or macrophage cells are capable of interacting with various pattern recognition receptors (PRRs) present on bacteria, viruses etc. and are the perfect candidates for the peptide epitopes considered here.18,19 The vaccine candidates or the epitopes on interacting with the surface TLRs are expected to elicit co-stimulatory signal for the development of mature; T-cells from naive T-cells specific to the infection.20 The interactions of individual peptide epitopes to TLRs were evaluated to narrow down candidate epitopes for final vaccine development. Altogether 18 docking experiment were performed by using the online platform of MDockPep between TLR2 (PDB ID: 3A79), TLR4 (PDB ID: 3VQ2) and TLR5 (PDB ID: 3J0A) with identified B-cell epitopes (SBepi 6) each from spike glycoprotein and RdRp (RBepi1-6).21 The binding energy values for each interactions were analyzed to assess the binding mode between the interacting interfaces. The binding energy value or Gibbs free energy (ΔG) is an important thermodynamics variable deciding the interaction of protein-epitope complex in the cellular condition.22 These values becomes the founding base for selection of suitable epitopes for final prophylactic vaccine development. The binding energy values of peptide with surface TLRs are shown in supplementary Table 3.
Table (3):
Binding energy values of peptide with surface TLRs.
No. |
TLR + B-cell epitope |
Binding energy (kcal.mol-1) |
|---|---|---|
1 |
TLR4 -VDTDLTKPYIKWDLLKYDFT |
-297.1 |
2 |
TLR5 – VDTDLTKPYIKWDLLKYDFT |
-292.7 |
3 |
TLR4 – FSNVTWFHAIHVSGTNGTKR |
-291 |
4 |
TLR2 – VDTDLTKPYIKWDLLKYDFT |
-288.2 |
5 |
TLR4 – FPNITNLCPFGEVFNATRFA |
-286.9 |
6 |
TLR2 – FSNVTWFHAIHVSGTNGTKR |
-274.8 |
7 |
TLR5 – FSNVTWFHAIHVSGTNGTKR |
-265.4 |
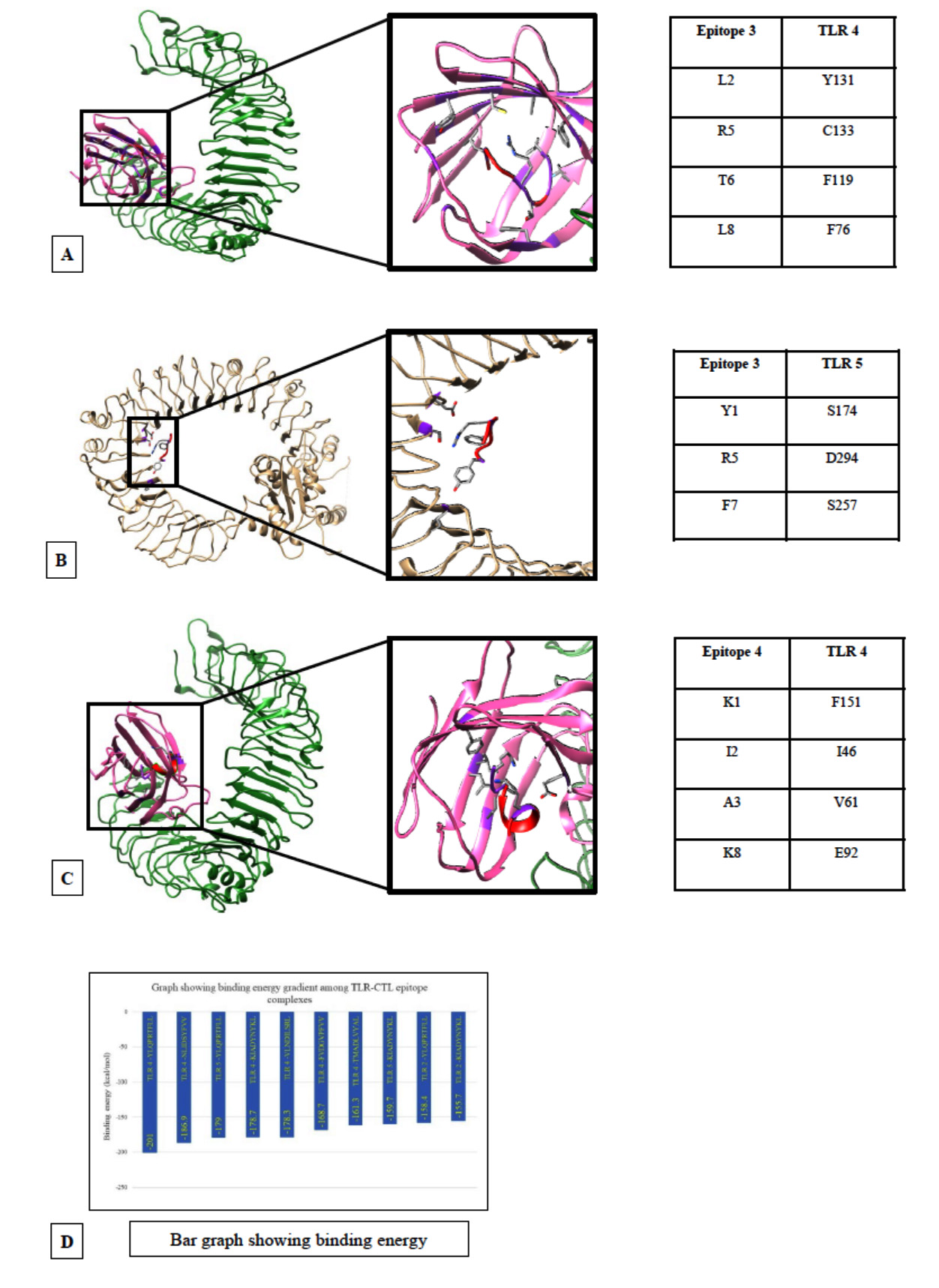
Fig. 3. Docking models of TLRs with CTL epitopes. (A) TLR4 docked with CTL epitope YLOPRTFLL obtained from spike protein. (B) TLR5 docked CTL epitope NLIDSYFVV obtained from RdRp. (C)TLR4 docked with CTL epitope KIADYNYKL obtained from spike protein of SARS-CoV-2. (D) Graph showing the binding energy.
The docked model of TLR4 and 269YLQPRTFLL277 in Fig. 3A represent the predominant role of hydrophobic interaction where CTL epitope was found to be well buried in the hydrophobic pocket mediated by the MD-2. The most important hydrophobic interactions which are exhibited by the complex are between leucine rich region of epitope L2, T6, L8 and Y131, F119, F76 of MD-2 respectively.
In TLR4-417KIADYNYKL425, the strong interaction is primarily mediated by salt-bridge between epitope Lys8 and TLR4 Glu92 further supported the hydrophobic interactions within the MD-2 cup (Fig. 3C)
The third docked model also represents salt bridge between epitope Asp3 and TLR Arg90. Additional hydrophobic interactions between epitope Val2and TLR4 Phe126 were observed. The TLR4 and 123 TMADLVYAL131, protein-peptide interaction is mainly mediated by hydrophobic residues.
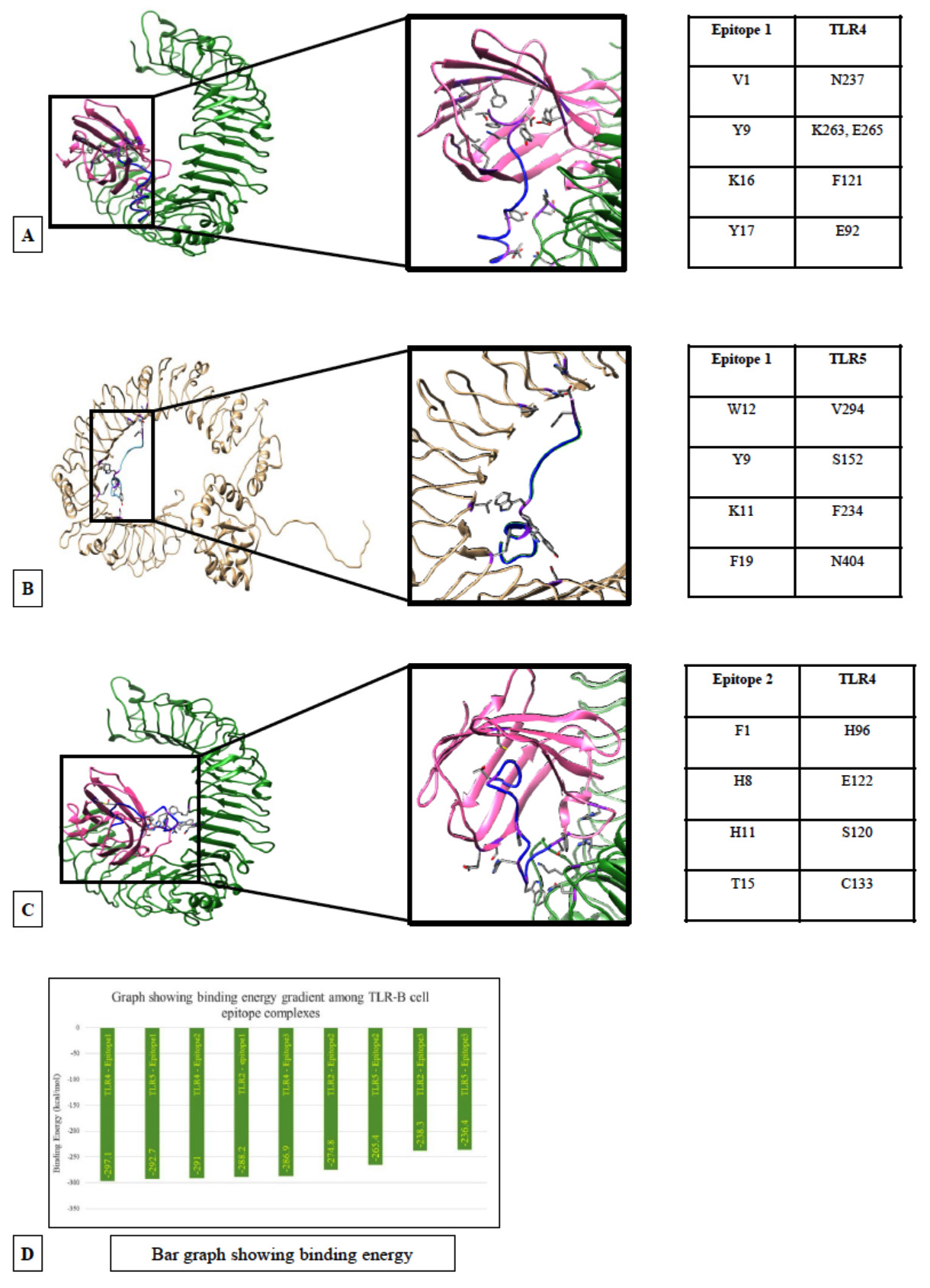
Fig. 4. Docking models of TLRs with B cell epitopes. (A) TLR4 docked with epitope VDTDLTKPYIKWDLLKYDFT obtained from RdRp. (B) TLR5 docked B cell epitope VDTDLTKPYIKWDLLKYDFT obtained from RdRp. (C)TLR4 docked with B cell epitope FPNITNLCPFGEVFNATRFA obtained from spike protein of SARS-CoV-2. (D) Graph showing the binding energy
TLR4 is also found to have high binding affinity with the three B-cell epitopes taken into the vaccine construct namely, 257VDTDLTKPYIKWDLLKYDFT276 (257R276)(Fig 4B), 59FSNVTWFHAIHVSGTNGTKR78(59S78), 329FPNITNLCPFGEVFNATRFA348(329S348), (Fig. 4C). The average binding energy of the epitopes with TLR4 is (-291.667+ 5.13) kcal.mol-1. The high negative binding energy indicates that a pro-inflammatory response is likely to be triggered by the interaction.
In TLR4 and 257VDTDLTKPYIKWDLLKYDFT276 (257R276)docked model Fig. 4B; (binding energy: -297.1 kcal.mol-1) the structure is stabilized by hydrophobic interactions and hydrogen bonds. Of them, the hydrogen bond between Y9 of the epitope and K263, E265 of the TLR4 is important and also that between Y19 of the epitope and E92 of MD-2. The inner hydrophobic cup of the MD-2 is surrounding the C-terminal of the epitope. Overall the role of hydrophobic residues for interaction of TLR4-MD2 with bacterial endotoxin were established earlier.24
In TLR4-59FSNVTWFHAIHVSGTNGTKR78(59S78), docked model, the binding energy is -291 kcal.mol-1 and the interaction is stabilized by an electrostatic bond between His8 of the epitope and E122 of MD-2.
TLR4 – 329FPNITNLCPFGEVFNATRFA348 (329S348) docked model have a binding energy of -286.9 kcal.mol-1 mediated by ionic to hydrophobic interaction tabulated in Fig 4C.
Docking with TLR5: TLR5 shows very good interaction with the T-cell epitopes 269YLQPRTFLL277 (269S277) (-179.7kcal.mol-1),417KIADYNYKL425 (417S425) (-159.7kcal.mol-1) and B-cell epitope 257VDTDLTKPYIKWDLLKYDFT276 (257R276) (-292.7kcal.mol-1) (Fig. 4B) with respective binding affinity values of -179.7kcal.mol-1, -159.7kcal.mol-1 and (-292.7kcal.mol-1 . The TLR5-YLQPRTFLL is mainly stabilized by hydrophobic interaction supported by electrostatic bonding between R5 of epitope and D295 of TLR5. In case of the B-cell epitope, hydrophobic bonds predominantly stabilize the binding with CTL epitopes.
Docking with TLR2: TLR2 has also made interaction with CTL and B-cell epitopes, but the binding energy in all the cases indicate weak binding affinity.
Overall, the approach screened the high and moderately interacting peptide epitopes to balance the pro-inflammatory response in the recipient body post-vaccination.
Multi-epitope vaccine construction
For designing a composite multiepitope vaccine, regions of RdRp and spike protein harbouring candidate B-cell, Th-cell and Tc-cel epitopes were mapped. The regions containing atleast one potential B-cell epitopes, five potential Th-cell epitopes and two Tc-cell epitopes from both the proteins were selected for composite antigen construction. The portion of RdRp bearing Tc and Th-cell epitopes and a B-cell epitope along with some amino acid residues flanking the ends of this region was selected for the vaccine construct. Similarly, the portion of the spike protein carrying two B-cell epitopes and Tc and Th-cell epitopes along with some amino acids flanking the ends, was selected. Finally, linker peptides like GPGPG were also introduced between individual epitope domains in order to give a conformational flexibility to the structure of final recombinant antigen as vaccine molecule. Interestingly both Repi3 and Sepi3 are peptides with high conservancy (>8). On the contrary Sepi1 is an epitope, exclusive for SARS-CoV-2 with less conservancy (<5).
Antigenicity and allergenicity of recombinant antigen
The recombinant antigen was thereby tested for its antigenicity in two different servers which predicted a moderate level 0.4532 in Vaxijen 2.0 and 0.641587 in ANTIGENpro. Further, the recombinant antigen sequence were tested for allergenicity using servers like AllerTop and AllergenFP which predicted the construct to be a probable non-allergen. The protein nearest to this vaccine construct was genome polyprotein of Triticum mosaic virus as predicted by Allertop. According to AllergenFP, the nearest protein was Sodium/ potassium-transporting ATPase subunit alpha-4 of human and a non-allergen. It has the highest Tanimoto index of 0.8.
Prediction of physico-chemical parameters for recombinant antigen as multiepitope vaccine
Physio-chemical parameters crucially determine stability, transportation, and immunogenicity of recombinant immunogens Safavi et al.25,26 Exploiting ProtParam physicochemical features of the composite peptide was determined. The molecular weight and isoelectric point for the 619 amino acid long antigen was predicted to be 70.67 kDa and 5.77 respectively. With an instability index computed to be 27.43, the immunogen apparently is stable. The projected half-life in mammalian reticulocytes (in vitro) and E. coli (in vivo) were computed to be 0.8 and 10 h respectively. Kyte-Doolittle hydropathy plot (FigSX) suggested regions of high polarity throughout the antigen which was further corroborated by rand average hydropathicity (GRAVY) and aliphatic index of -0.188 and 79.63 respectively. The polarity Grantham plot suggested predominance of hydrophilic stretches in the antigen with a predicted solubility of 0.684 (ProSOL) in E. coli upon over expression. Post-translational modification often affect the effective immunogenicity of DNA vaccines. An intricate protein motif analysis using Motif Scan, predicted absence of glycosylation sites in the antigen indicating maximum accessibility of the antigen to immune cells. Cumulatively, analysis of physicochemical parameters further substantiated prospect of the composite multiepitope immunogen with respect to stability and accessibility.
In silico cloning and vaccine optimization
Bacterial expression construct: The vaccine construct, an amino acid sequence, was codon optimized and converted into a codon sequence using BackTranseq with codon optimization for Escherichia coli K12. The codon sequence of the restriction sites of the restriction enzymes NdeI and BamHI was added to the 5’ and 3’ terminal of the adapted codon sequence respectively. This yielded the total codon sequence of 1872bp. After performing in silico cloning of the adapted sequence in pET-16b expression vector by means of SnapGene tool, its solubility was checked upon overexpression in E.coli. The predicted solubitity was 0.684499.
DNA vaccine design
For DNA vaccine development, the codon optimization of the composite peptide was performed against Homo sapiens. In silico cloning was performed in pVax 1.0 after addition of HindIII and Xba1 site to the 5’ and 3’ terminal of the adapted codon sequence respectively. A secretory signal, comprising 33 amino acid (99 bases) of N Terminal sequence of Human growth hormone (HGH), as a secretory signal.
In-silico modelling of the recombinant antigen and molecular docking with TLRs
The recombinant protein designed were allowed to generate a three-dimensional model using independent platform of Swiss-model and I-Tasser.27,28 The models generated were validated by Ramachandran plot and I-tasser model found to have 98 % residues in allowed and partially allowed region of plot. The secondary structure organization of the model was validated by PsiPred secondary structure prediction software. The model of recombinant antigen highlighted with epitopes in red and blue colour is shown in Fig. 4.
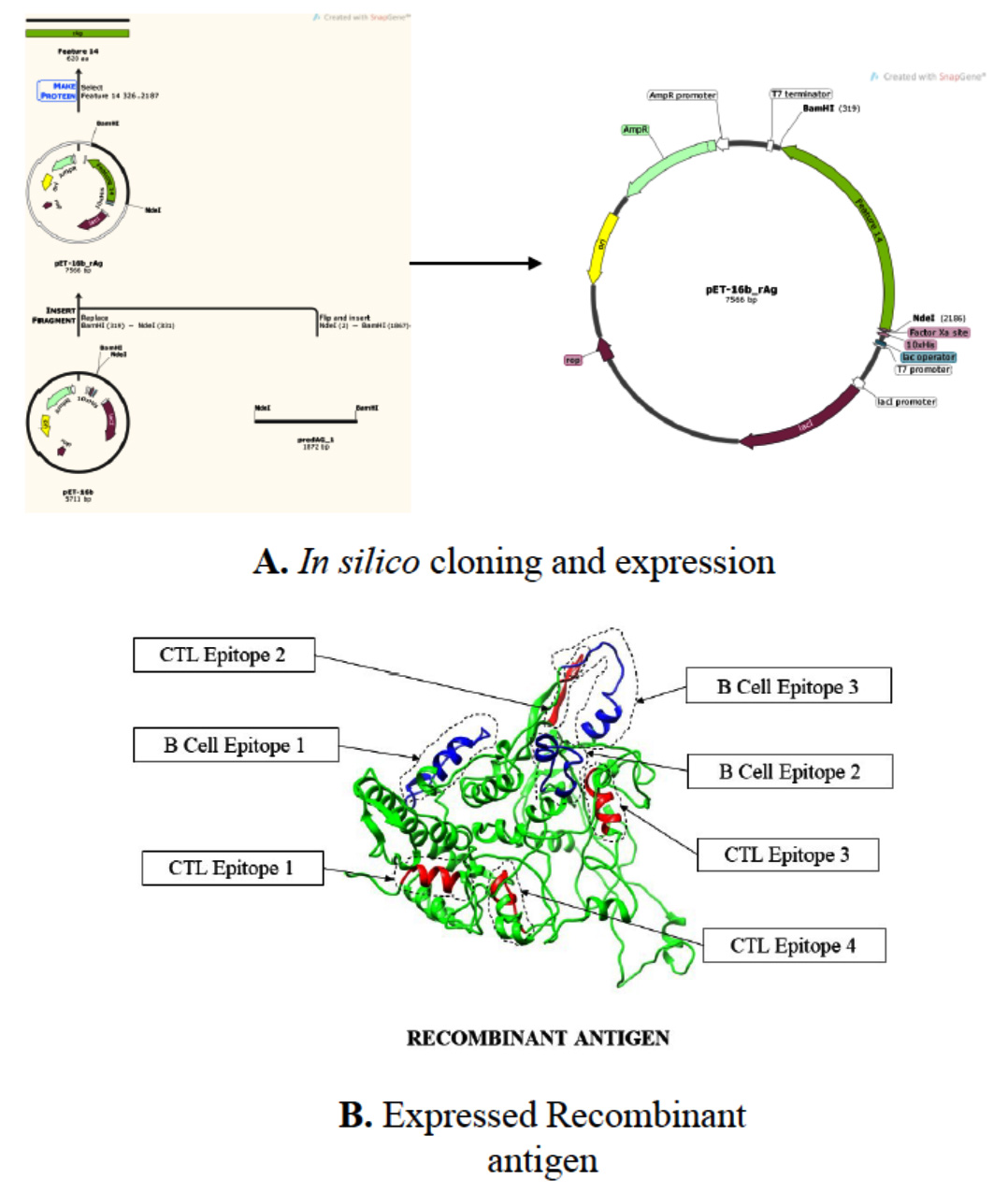
Fig. 5. A) in silico cloning and expression. B) Expressed recombinant antigen [Green Recombinant antigen Blue B-cell epitope Red CTL epitope].
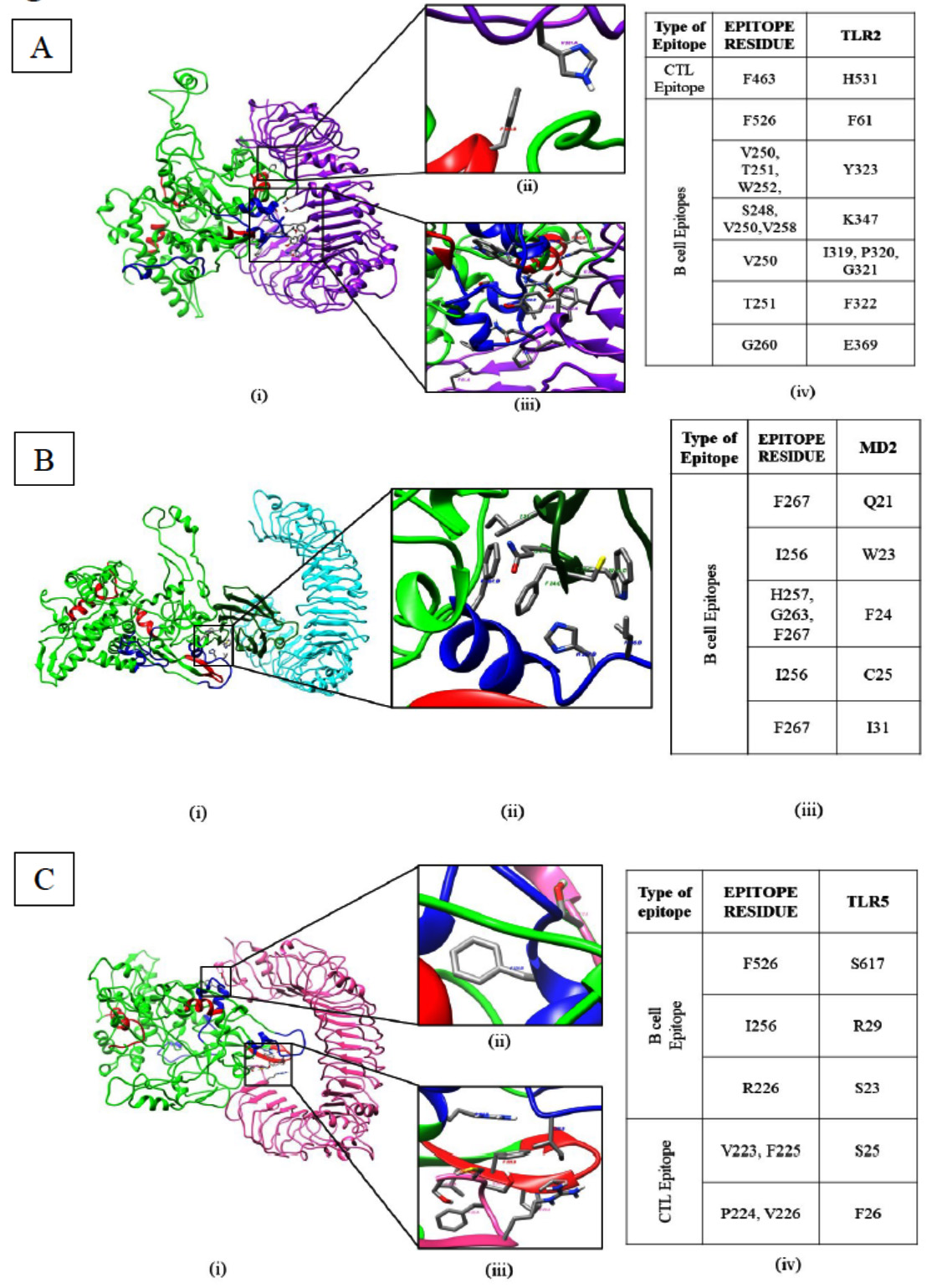
Fig. 6. Molecular docking of TLRs with RA. A) Docking of TLR2 with recombinant antigen. B) Docking of TLR4-MD2 with recombinant antigen. [(i) model (ii) interactions (iii) chart]. C) Docking of TLR5 with recombinant antigen[ (i) Model (ii, iii) interaction (iv) chart ] [Green Recombinant antigen Red CTL epitope Blue B-cell epitope Purple TLR2 Cyan TLR4 Bottle green MD2 Pink TLR5].
The molecular docking of these recombinant antigen were performed against surface TLRs using Haddock to analyze their effectivity against surface TLRs. Docking data shows a very good interaction of recombinant antigen with all three TLRs where hydrophobic bonds prevalently stabilize the molecular interactions. Interestingly, TLR4 interacts with recombinant antigen only through MD2 protein and support the role of MD2 in TLR4 interaction with other antigens. A detailed residual interactions between TLRs and recombinant antigen has been tabulated and represented in Fig. 6.
The evolution and arrival of highly infectious new genetic variants of SARS-CoV-2 seems to have a role in affecting efficacy of available vaccines and the scientific world are attempting several modification in immunoinformatics approaches to predict newer vaccine candidates in a limited period of time. In this study of in silico vaccine design, we aimed at using three surface TLRs namely TLR2, TLR4 and TLR5 as a means of selecting the most potent epitopes, as they can sense microbial pathogens by identifying pattern-recognition receptors (PRRs) present on the surfaces of them.29 The spike protein and the RNA dependent RNA polymerase was thoroughly scanned for conserved immunogenic epitopes. Due to the recent rise in mutated variants of SARS-CoV-2, it was a necessary strategy to aim at a protein sequence which should be immunogenic and more conserved in most of the mutants. That may reduce the need for newer vaccine candidates for each variant. Earlier gene content studies identified that surface spike glycoprotein is more prone to mutagenesis (61%) than endogenous proteins like RdRp (17%).30 The multiple sequence alignment of proteins among the three coronaviruses namely SARS-CoV-2, SARS-CoV and MERS-CoV in question, represents the more conserved nature of RdRp rather the spike protein. Incorporating epitopes from endogenous proteins in recombinant antigen may provide a protection against newly evolved genomic variants of SARS-CoV-2.7
The search for epitopes in both spike and RdRp proteins returned us with a cluster of epitopes ranging from intermediate to conserved residual content. The conserved epitopes were contributed mainly from the RdRp. Instead of joining each and every epitope end-to-end with linkers, we picked up portions from the protein of our choice. The resulting two portions; one from RdRp and the other from spike protein, were joined together by linker. The sequence thus obtained, was reverse translated computationally and the most probable codon sequence was obtained, administering codon bias for bacteria E. coli. The vaccine is designed to be a peptide vaccine containing the antigenic regions of the virus for exhibition to the human immune system with the hope of immunizing the humans. We had chosen E. coli as our expression system for ease of genetic manipulation. The recombinant antigen was thoroughly checked for any sign of allergenicity without compromising the antigenicity as well as the efficacy. Our repeated checks proved fruitful and the antigen thus designed was non-allergen but immunogenic.
In the human immune system, the T-cells and B-cells do the bulwark in protecting the body against any sort of pathological infection. With this in mind and the knowledge of memory T- and B-cell development, the epitopes were mapped. It is well established about the role of PRRs in starting off with the innate immunity, the first line of defense against pathogens; and TLRs play a vital role in this pathogen recognition phase.20 TLRs are distributed both on the cell surface of APCs as well as within lysosomes; LRR region being inside the lysosomal vesicle. These intracellular TLRs are designed to recognize PAMPs of nucleic acids in nature and of viral origin. on the other hand, cell surface TLRs recognize PAMPs of peptide nature or liposaccharides.31 From this standpoint, we performed the docking of individual epitopes with the three shortlisted surface TLRs which exhibited high binding energy with the epitopes. The magnitude of binding energy between selected epitopes and TLRs were used to design the recombinant antigen as a vaccine candidate. Another criteria of binding potentiality for recombinant antigen with the TLRs were analyzed for good to moderate level of binding ensuring the efficacy as hypothesized.
However, it should be noted that this is wholly a computational and conclusive vaccine candidate shortlisting demands extensive experimental works and data.
Additional file: Additional Table S1-S2. Additional Figure S1 and S2.
ACKNOWLEDGMENTS
None.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
AUTHORS’ CONTRIBUTION
All authors designed the experiments. NB and AD performed the experiments. KP, AB, JP and SH analyzed the data. AB and KP wrote the manuscript. All authors read and approved the final manuscript for publication.
FUNDING
This work was supported by the Department of Science and Technology (DST)-SERB grant (File Number: SRG/2020/002215) awarded to author KP and (File Number: SRG/2020/000702) awarded to author AB. The work is also supported by seed grant of authors AB, JP and SH.
ETHICS STATEMENT
Not applicable.
AVAILABILITY OF DATA
All datasets generated or analyzed during this study are included in the manuscript and/or in the supplementary files.
- Hui DS, Azhar EI, Madani TA, et al. The continuing 2019-nCoV epidemic threat of novel coronaviruses to global health – The latest 2019 novel coronavirus outbreak in Wuhan. China Int J Infect Dis. 2020;91:264-266.
Crossref - Wang C, Horby PW, Hayden FG, Gao GF. A novel coronavirus outbreak of global health concern. Lancet. 2020;395(10223):470-473.
Crossref - Grifoni A, Weiskopf D, Ramirez SI, et al. Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans with COVID-19 Disease and Unexposed Individuals. Cell. 2020;181(7):1489-1501,e15.
Crossref - Du L, He Y, Zhou Y, Liu S, Zheng B-J, Jiang S. The spike protein of SARS-CoV – a target for vaccine and therapeutic development. Nat Rev Microbiol. 2009;7(3):226-236.
Crossref - Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell. 2020;181(2):281-292.
Crossref - Tahir Ul Qamar M, Rehman A, Tuslum M, et al. Designing of a next-generation multiepitope based vaccine (MEV) against SARS-COV-2: Immunoinformatics and in silico approaches. PLoS One. 2020;15(12):e0244176.
Crossref - Mahase E. Covid-19: How many variants are there, and what do we know about them? BMJ. 2021;374:n1971.
Crossref - Siegrist CA, Aspinall R. B-cell responses to vaccination at the extremes of age. Nat Rev Immunol. 2009;9(3):185-194.
Crossref - Mishra KP, Singh AK, Singh SB. Hyperinflammation and Immune Response Generation in COVID-19. Neuroimmunomodulation. 2020;27(2):80-86.
Crossref - Faisal AM, Imtiaz SH, Zerin T, Rahman T, Shekhar HU. Computer-aided epitope design as a peptide vaccine component against Lassa virus. Bioinformation. 2017;13(12):417-429.
Crossref - Shastri N, Serwold T, Gonzalez F. Presentation of endogenous peptide/MHC class I complexes is profoundly influenced by specific C-terminal flanking residues. J Immunol. 1995;155(9):4339-4346. PMID: 7594593
- Reimann J, Schirmbeck R. Alternative pathways for processing exogenous and endogenous antigens that can generate peptides for MHC class I-restricted presentation. Immunol Rev. 1999;172(1):131-152.
Crossref - Schirmbeck R, Reimann J. Alternative processing of endogenous or exogenous antigens extends the immunogenic, H-2 class I-restricted peptide repertoire. Mol Immunol. 2002;39(3-4):249-259.
Crossref - Testa JS, Philip R. Role of T-cell epitope-based vaccine in prophylactic and therapeutic applications. Future Virol. 2012;7(11):1077-1088.
Crossref - Panina-Bordignon P, Tan A, Termijtelen A, Demotz S, Corradin G, Lanzavecchia A. Universally immunogenic T cell epitopes: promiscuous binding to human MHC class II and promiscuous recognition by T cells. Eur J Immunol. 1989;19(12):2237-2242.
Crossref - Paul S, Sidney J, Sette A, Peters B. TepiTool: A Pipeline for Computational Prediction of T Cell Epitope Candidates. Curr Protoc Immunol. 2016;114(1):18 9 1- 9 24.
Crossref - Jespersen MC, Peters B, Nielsen M, Marcatili P. BepiPred-2.0: improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res. 2017;45(W1):W24-W29.
Crossref - Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev. 2009;22(2):240-273.
Crossref - Vijay K. Toll-like receptors in immunity and inflammatory diseases: Past, present, and future. Int Immunopharmacol. 2018;59:391-412.
Crossref - Jin B, Sun T, Yu XH, Yang YX, Yeo AE. The effects of TLR activation on T-cell development and differentiation. Clin Dev Immunol. 2012;2012:836485.
Crossref - Xu X, Yan C, Zou X. MDockPeP: An ab-initio protein-peptide docking server. J Comput Chem. 2018;39(28):2409-2413.
Crossref - Vangone A, Schaarschmidt J, Koukos P, et al. Large-scale prediction of binding affinity in protein-small ligand complexes: the PRODIGY-LIG web server. Bioinformatics. 2019;35(9):1585-1587.
Crossref - Palm NW, Medzhitov R. Pattern recognition receptors and control of adaptive immunity. Immunol Rev. 2009;227(1):221-233.
Crossref - Resman N, Vasl J, Oblak A, et al. Essential roles of hydrophobic residues in both MD-2 and toll-like receptor 4 in activation by endotoxin. J Biol Chem. 2009;284(22):15052-15060.
Crossref - Safavi A, Kefayat A, Mahdevar E, Abiri A, Ghahremani F. Exploring the out of sight antigens of SARS-CoV-2 to design a candidate multi-epitope vaccine by utilizing immunoinformatics approaches. Vaccine. 2020;38(48):7612-7628.
Crossref - Safavi A, Kefayat A, Abiri A, Mahdevar E, Behnia AH, Ghahremani F. In silico analysis of transmembrane protein 31 (TMEM31) antigen to design novel multiepitope peptide and DNA cancer vaccines against melanoma. Mol Immunol. 2019;112:93-102.
Crossref - Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. The I-TASSER Suite: protein structure and function prediction. Nat Methods. 2015;12(1):7-8.
Crossref - Waterhouse A, Bertoni M, Bienert S, et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46(W1):W296-W303.
Crossref - Pulendran B, Ahmed R. Immunological mechanisms of vaccination. Nat Immunol. 2011;12(6):509-517.
Crossref - Jungreis I, Sealfon R, Kellis M. SARS-CoV-2 gene content and COVID-19 mutation impact by comparing 44 Sarbecovirus genomes. Nat Commun. 2021;12(1):2642.
Crossref - Majewska M, Szczepanik M. The role of Toll-like receptors (TLR) in innate and adaptive immune responses and their function in immune response regulation. Postepy Hig Med Dosw. 2006;60:52-63.
- Sievers F, Higgins DG. Clustal Omega, accurate alignment of very large numbers of sequences. Methods Mol Biol. 2014;1079:105-116.
Crossref
© The Author(s) 2022. Open Access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License which permits unrestricted use, sharing, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.