


ISSN: 0973-7510
E-ISSN: 2581-690X
A total of 407 samples were collected from the western region of Saudi Arabia. Soil samples as well as dead larvae of Spodoptera littoralis (cotton leaf worm) were collected and examined for the presence of Bacillus thuringiensis. The bacterium was isolated using an acetate-selective enrichment medium and plating. The isolates that were collected by the above-mentioned protocol were identified by using a combination of both microscopic examinations as well as the analysis of 16S rRNA genes through DNA sequencing of PCR products. A total of 22 confirmed isolates of Bacillus thuringiensis were identified for the purpose of this study. Collectively, these confirmed isolates make up 4.6% of all soil samples and 6.6% of all dead larvae samples that were collected. Although B. thuringiensis was not found to be abundant in soil habitats of The Makkah Province, our results suggest that the bacterium is the part of the indigenous microflora of the area that we explored in this study. Preliminary results show that the partially purified crystal protein of some of the isolates exhibits demonstrable activity against three human cancer cell lines: Hep G2 (human liver carcinoma cell line), HeLa cells (human uterine cervix cancer cell line), and Vero cells (lung cancer Cell line).
Bacillus thuringiensis, 16srRNA, Hep; HeLa, Vero.
It is well known that cancer is one of the leading causes of death worldwide. In 2004 cancer was responsible for 7.4 million deaths worldwide and, as per the prediction of the World Health Organization, this number will increase to 12 million deaths by the year 2030. Although the Kingdom of Saudi Arabia has one of the lowest incidences of cancer in the world, the number of reported cases of cancer is rising rapidly. Even though cancer is regarded as one of the leading causes of death in the world, it is possible to avoid or prevent nearly 30% of all cancer-related deaths (Almas et al., 2015). B. thuringiensis (Bt), a ubiquitous gram-positive spore-forming bacterium, forms parasporal proteins during the stationary phase of its growth (Osman et al., 2015). Recent studies focused on analyzing selective human cancer cell-killing activities have resulted in the discovery of a new category of Bt parasporal proteins called parasporins (Almas et al., 2015). Pathogenic microbes targeting vertebrate and invertebrate organisms are known to produce a variety of proteinaceous toxins some of which have the ability to pass through plasma membranes. Most of the pore-forming toxins bind to microdomains present on the eukaryotic cell surfaces, called lipid rafts, via specific receptors (Manes, et al., 2003). Subsequent to this binding the hydrophilic toxins become inserted into the cellular membrane and form aqueous pores constructed from physically stable toxin oligomers. Interest in toxins that target lipid rafts has dramatically increased off late not only because of their mode of action but also for the possibility of using them as molecular tools for investigating and visualizing the elusive membrane lipids rafts (Waheed et al., 2001). A majority of the crystal (Cry1) proteins produced by B. thuringiensis are thought to form pores in the plasma membranes of epithelial cells present in the insect midgut (Schnepf et al.,1998 ; de Maagd et al., 2003). The toxins show selective insecticidal actions against one or more insects but not against other organisms. Several hundred genes encoding for Cry proteins have been isolated from insecticidal B. thuringiensis strains and some of these are now being used for pest control in agriculture (Elmenofy et al., 2014). Interestingly, several strains that exhibit cytotoxic activity towards mammalian cells have been found among non-insecticidal and non-hemolytic B. thuringiensis strains (Ohba, and Aizawa, 1986). Several novel Cry proteins, designated as parasporin-1,-2,-3-4,-5 and -6 have recently been identified (Mizuki, et al., 2000; Ito, et al., 2004; Katayama, et al., 2005; Yamashita, et al., 2005; Saitoh, et al., 2006) and it is of great interest that some of these proteins exhibit cytotoxic activities against specific human carcinoma cells. In particular, it has been clearly demonstrated that parasporin-2 preferentially kills cancer cells in the slices of liver and colon cancer tissues while leaving the normal cells unaffected (Ito, et al., 2004; Kuroda et al., 2013). Parasporin-2 (Cry46Aa1) was originally purified from the parasporal inclusions of the B. thuringiensis strain A1547 as a cytolytic protein that demonstrated activity against human leukemic T cells (Ito et al., 2004). The cytotoxicity of the toxin varies considerably between different cell types. For example, it is highly cytotoxic toward HepG2 (human hepatocyte cancer) cells but considerably less toward HC (human normal hepatocyte) and HeLa (human uterine cancer) cells. The toxin appears to act by binding specifically to the plasma membrane of susceptible cells and increasing their membrane permeability followed by the induction of significant changes in the cytoskeleton and organelle morphologies (Kitada, et al., 2006). Although parasporin-2 is thought to be a selective membrane pore-inducing toxin, the nature of its actions toward the cellular membrane has not yet been characterized. Kitada et al., 2006 discovered that parasporin-2 forms SDS-resistant oligomers (200 kDa) on the membrane of target cells. Their study also revealed that oligomerization of the toxin was related to permeabilization of the plasma membrane and that the phenomenon was dependent on both membrane proteins as well as the lipid bilayer of the target cell. The toxin monomers were observed to be bound peripherally to the lipid rafts present on the plasma membrane of the target cell. Our study aimed to screen and identify anticancer agents from bacteria that have been isolated from the western region of Saudi Arabia.
Isolation of B. thuringiensis from soil
A total of 257 soil samples were collected from Makkah City (urban soil), the Al-Jamoom area which is located 40 km off the eastern outskirts of Makkah (agricultural soil), the Al-Konfodhah City located 350 km south-west of Makkah City (beach sand) and from Jizan City which is situated 685 km south-west of Makkah City (desert sand). The samples were collected from a depth of 2 to 5 cm below the surface using a shovel. Each soil sample was placed inside a plastic bag at ambient temperature. B. thuringiensis strains were isolated from the collected soil samples using L B and T3 agar medium as per the method described by Travers et al., (1987).
Isolation of B. thuringiensis from dead larvae
A total of 150 samples of dead cotton leafworm larvae (Spodoptera littoralis; locally known as the alfa alfa leaf worm) were collected from the Makkah area. The dead larvae were collected using sterile forceps and placed in sterile plastic screw-top bottles. Subsequent to the collection, each dead larva was crushed in a sterile crucible and added to a tube containing 9 mL sterile phosphate buffered saline (PBS). The suspension was heated at 80°C for 3 min and then centrifuged at 8000 rpm for 5 min. A 50 ¼L aliquot of the supernatant was spread on LB agar plates and incubated overnight at 30 °C (Osman et al., 2007).
Preparation solubilisation and activation of parasporal protein
Sporulated Bt cultures was treated with 1 M NaCl to osmotically lyse the bacterium to release the spore and crystals. The spore-crystal mixture was harvested by centrifugation at 13,000 g for 5 minutes, washed once with NaCl and twice with ice-cold water. The sporecrystal mixture was resuspended in Tris/KCl buffer (pH 7.5) before storing at -20°C. The parasporal protein was separated from spores by ultracentrifugation of the spore-crystal mixture at 25000 g, 4°C for 16 hours on a discontinuous sucrose density gradient of 67, 72 and 79% (w/v) in Tris/KCl buffer (pH 7.5) (Thomas and Ellar 1983). The parasporal protein was solubilised in sodium carbonate buffer (pH 10.5) containing 10 mM DTT and activated with trypsin (1 mg/mL) for 1 hour at 37°C. The activated parasporal protein was desalted and concentrated using Amicon® Ultra-4 centrifugal filter (Milipore). Protein concentration was estimated using the method of Bradford (Bradford, 1976) using bovine serum albumin (BSA) as standard.
Preparation of Chromosomal DNA from Bacterial Isolates
Genomic DNA was isolated as per the protocol described by the manufacturers (Promega, USA). A single bacterial colony was transferred into a loosely capped 10 mL tube containing 5 mL of LB agar-pate. The culture was incubated overnight at 37°C with vigorous shaking. Of the culture, 1 mL was transferred to a micro centrifuge tube and centrifuged at 13,000-16,000 xg for 2 min so as to pellet the cells. To this 240 µL of 10 mg/mL lysozyme was added. The solution was gently pipetted to ensure complete mixing. The purpose of this pretreatment was to weaken the cell wall so that efficient cell lysis can take place. The sample was then incubated at 37°C for 30–60 min followed by centrifugation at 13,000–16,000 xg for 2 min. The supernatant was removed and discarded. Of the nuclear lysis solution, 600 µL was added to the pellet and the solution was gently pipetted so as to ensure that the cell pellet was properly resuspended. The samples were then incubated at 80 °C for 5 min to facilitate cell lysis and then cooled to room temperature. Of an RNAase solution (10 mg/mL), 3 µL was added to the above and the tube was inverted 2-5 times for complete mixing. The tubes were then incubated at 37°C for 30 min followed by cooling the sample to room temperature. Of protein precipitation solution, 200 µL was added to the RNAase-treated cell lysate and vortexed vigorously at high speed for 20 s for mixing the protein precipitation solution with the cell lysate. The sample was incubated on ice for 5 min and centrifuged at 13,000 xg for 3 min. The supernatant was transferred to a clean 1.5 mL micro centrifuge tube containing 600 µL of room temperature isopropanol, gently mixed by inversion until the thread-like strands of DNA form a visible mass and centrifuged at 13,000-16,000 xg for 2 min. The supernatant was carefully discarded by decanting and the tube was dried on a clean absorbent paper. Of 70% ethanol, 600 µL at room temperature was added and the tube was gently inverted several times to wash the DNA pellet. This was then centrifuged at 13,000-16,000 xg for 2 min and the ethanol removed by careful aspiration. The tube was drained on a clean absorbent paper and the pellet was allowed to air–dry for 10–15 min after which the genomic DNA was dissolved in 100 µL of DNA rehydration solution and stored at –-20°C till required (EL-Ghareeb et al., 2012).
PCR of 16SRNA
Polymerase Chain Reaction is a procedure that allows for rapid detection of the presence or absence of a target DNA sequence in any given pool of genetic material (Assaeedi et al., 2012). As a molecular confirmatory test, the 42 Bacillus-like isolates that had previously been investigated under the microscope were used for PCR of the 16S rRNA gene as per the protocol described previously (Mohamed et al., 2006; Abulreesh et al., 2012). PCR amplification was then followed by sequencing analysis of the amplicon. Primers used for the PCR procedure were synthesized by Bioneer, South Korea. The primer sequences used for the above-mentioned purpose were as follows:
F-5’-AGTTTGACTCTGGCTCAG-3’ and R-5’-TACGGCTACCTTGTTACGACTT-3’
The composition of the PCR reaction mixture (50 µL total volume) was as follows: (a) 200 µM of each dNTP, (b) 0.5 µM primers (F and R), (c) 10 mM Tris-HCl pH 8.3, (d) 1.5 mM MgCl2, (e) 50 mM KCl, (f) 2.5 units Taq polymerase (Applied Biosystems, UK) and, (g) 100 ng DNA template. The PCR thermal cycling parameters used for amplification of DNA (primers: F1 and R1) were as follows: 94°C for 3 min followed by 30 cycles of 94°C for 1 min, 54°C for 1 min and 72°C for 1.5 min. The efficacy of the PCR reaction was assessed by analyzing 20 µL of the product by agarose gel electrophoresis (1% gel). Ethidium bromide (0.5 mg/mL) was used for the purpose of DNA staining and a UV transilluminator was used for visualization. The amplified fragments were eluted and sequenced according to the protocol described by Sanger et al., (1977)
Cell culture and analysis of anticancer activity and selectivity
Three human cancer cell lines were used for the purpose of studying anticancer activity. These three cell lines were (a) the human cervical cancer HeLa cells (human uterine cervix cancer cell line), (b) the Vero cell line (human lung cancer cell line), and (c) Hep G2 (human liver carcinoma cell line). These cell lines were obtained from the American Type Culture Collection (ATCC). The cells were grown and maintained in either RPMI1640 or DMEM growth medium (Lonza) supplemented with 10% FBS (Lonza), and 1% penicillin /streptomycin (Lonza). In a 75-mL tissue culture flask (Santa Cruz), the cell lines were incubated at 37°C in a humidified incubator containing 5% CO2 in the atmosphere. The growth medium of the cells was routinely changed once every two days. At confluence, the cells were split 1 to 3 and passaged as per routine protocol. The cells that had grown to approximately 90% confluence were detached from the substratum using 0.25% Trypsin–EDTA (Lonza) before being washed and re-suspended in the Complete Medium. For all the three cell lines, 2.5×104 cells were plated in triplicate in a 96-well plate (Santa Cruz) and incubated overnight at 37°C in a humidified 5% CO2 incubator. The plated cells were then treated with parasporal proteins been partially purified from eight bacterial isolates (100ug/ml) for of 24 h at 37°C. At the end of the treatment, proliferation and cytotoxicity assays were carried out to quantify the viable cell number. This was done by using the Cell Counting Kit-8 (CCK-8) assay procured from Sigma. Cell Counting Kit-8 (CCK-8) is a sensitive colorimetric assay for determining the number of viable cells present in proliferation and cytotoxicity assays. For further conformation, the cell proliferation test was also analyzed using the MTT assay (BioMatik).
Cytotoxicity tests
A Cell Counting Kit-8 (CCK-8) assay (Sigma Chemical Co., St. Louis, MO) were used to measure the cytotoxicity of different 8 treatment on HeLa, Hep G2 and Vero cell line. Where The CCK-8 assay was used to measure cytotoxicity (Ishiyama et al., 1996), which are based on the conversion of a water-soluble tetrazolium salt, 2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2Htetrazolium, monosodium salt (WST-8), to a water-soluble formazan dye upon reduction by dehydrogenases in the presence of an electron carrier. HeLa, Hep G2 and Vero cell (5 x104 cells/ml) were grown in 96-well plates for 48 h and treated then treated with parasporal proteins been partially purified from eight bacterial isolates (100 ul/ml of trypsin activated protein) After 24 h, the cell were washed, and the extent of cell growth was assessed using a CCK-8 assay. CCK-8 solution (10¼l) was added to each well, followed by incubation for 2 h at 37°C. The absorbance at 450nm was determined by a multiplate reader. Cell viability was expressed as a percentage of that of the control (untreated) cells. For each concentration of treatment, mean values of the mean absorbance rates from eight wells were calculated.
MTT assay for cell proliferation
Cell proliferation was determined by MTT assay according to (Alley et al., 1988; Meky et al., 2001). In brief, HeLa cells, Hep G2 and Vero cell line in logarithmic phase were seeded into 96-well plate at 1×104cells/well followed by incubation at 37°C for 24 h for attachment and then treated with (100ug/ml) for 24h, respectively. 20 ¼L of 5 mg/mL MTT dye was added to each well and incubated at 37°C for 4 h. Then the supernatant was discarded and purple-coloured precipitates of formazan were dissolved by gently shaking for 10 min in 150 ¼L of dimethyl sulfoxide (DMSO). After complete dissolution, absorbance (A) was measured at 490 nm on a microplate reader. The effect of different treatment on growth inhibition was assessed as the percentage of inhibition in cell growth. Background absorbance of the medium in the absence of cells was subtracted. Percent viability was calculated as [value of drug-treated group (A)/control group (A)] × 100%. Each assay was carried out three times, and the results were expressed as the mean (± SEM). Similar results were observed in at least three independent experiments.
Isolation of B. thuringiensis from soil and insect samples
A total 257 soil samples and 150 samples of dead cotton leaf worm larvae were collected from various areas in The Makkah province of western Saudi Arabia. All collected samples were examined and analyzed for the presence of B. thuringiensis. A total of 205 Bacillus isolates were recovered from the 257 soil samples that were collected for this study. However, based on the production of parasporal inclusions, only 12 of the 205 isolates could be confirmed as belonging to the B. thuringiensis species (Table 1). Along similar lines, of the 117 Bacillus isolates that were recovered from the dead larvae, only 10 isolates were found to produce parasporal inclusions. The B. thuringiensis index was calculated as the number of B. thuringiensis isolates recovered divided by the total number of Bacillus isolates examined.
Table (1):
Isolation of B. thuringiensis in soil and dead larvae samples from Western region of Saudi Arabia.
Location |
No. of soil samples |
No. of soil samples with Bt |
No. of Bacillus colonies isolated |
No. of Bt isolates |
Bt Index |
|---|---|---|---|---|---|
Makkah |
79 |
3 |
68 |
3 |
0.044 |
Al-jamoom |
56 |
2 |
41 |
2 |
0.048 |
Qunfudah |
34 |
1 |
27 |
1 |
0.37 |
Jezan |
23 |
2 |
18 |
2 |
0.11 |
Taaf |
48 |
3 |
37 |
3 |
0.081 |
Jeddah |
17 |
1 |
14 |
1 |
0.071 |
Total |
257 |
12 |
205 |
12 |
0.058 |
Location |
No. of dead larvae samples |
No. of larvae samples with Bt |
No. of Bacillus colonies isolated |
No. of Bt isolates |
Bt Index |
Taaf |
61 |
4 |
51 |
4 |
0.078 |
Jazan |
47 |
3 |
39 |
3 |
0.076 |
Al-jamoom |
42 |
3 |
27 |
3 |
0.11 |
total |
150 |
3 |
117 |
10 |
0.085 |
Identification of isolates by PCR amplification of the 16SrRNA gene
PCR amplification of the 16SrRNA gene of 42 isolates was performed using a single pair of primer. As a molecular confirmatory test, the 16SrRNA sequences of 42 isolates were PCR amplified and sequenced. (Fig. 1), followed by sequencing as a molecular confirmatory test. A 1.45 kb fragment of the 16S rRNA gene was amplified using the primer pair described previously in Methods section. The amplified fragment was sequenced so as to obtain the nucleotide sequence of the entire 1.45 kb amplicon. The NCBI-BLAST program was used to analyze the sequences obtained and homology search revealed that only 8 of the 42 isolates could be categorized as B. thuringiensis based upon 98% sequence identity.
 Fig. 1. Agarose gel electrophoresis of the PCR products of the 16S rRNA fragments for isolates 1 to 21. Panel B, M: 1 Kb DNA ladder; Panel A: isolates from 22 to 42 lane M: 1 Kb DNA ladder.
Fig. 1. Agarose gel electrophoresis of the PCR products of the 16S rRNA fragments for isolates 1 to 21. Panel B, M: 1 Kb DNA ladder; Panel A: isolates from 22 to 42 lane M: 1 Kb DNA ladder. Analysis of anticancer activity and selectivity
Hep G2 cells were treated with 90 µL of toxin for a period of 24 h. CCK-8 results for cytotoxicity assays are reported in the terms of relative cell viability (%). All data were normalized to the control group which was designated as 100%. The results revealed that the toxin inhibited proliferation of Hep G2 cells. *P<0.05 and ** P<0.01 versus the control group (0 ¼M) (two-way ANOVA followed by Tukey’s post hoc test). (Fig 2 & 3) (Table 2 & 3).
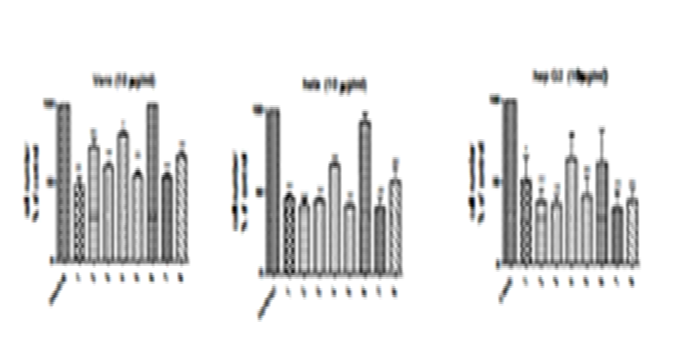 Fig. 2. A: There was a significant decrease in Vero cell number percentage. P > 0.05 after the treatment with 100 µg/mL of strain 4. While the treatment with strains 2, 3, 5, 7, and 8 (100 µg/mL) produced a significant decrease in Vero cell number percentage, P > 0.01 (cell number less than 50% with 1, 5, and 7). The treatment with strain 6 has no significant effect. B: There was a significant decrease in HeLa cell number percentage, P > 0.01 after treatment with 100 µg/mL of strain 1, 2, 3, 4, 5, 7, and 8. (cell number less than 50% except for strains 4 and 8). The treatment with strain 6 has no significant effect. C: There was a significant decrease in Hep G2 cell number percentage P > 0.05 after treatment with 100 ¼g/mL of strain 1. While the treatment with strains 2, 3, 5, 7, and 8 (100 µg/mL) produced significant decrease in Hep G2 cell number percentage P > 0.01 (cell number less than 50%). The treatment with strains 4 and 6 had no significant effect..
Fig. 2. A: There was a significant decrease in Vero cell number percentage. P > 0.05 after the treatment with 100 µg/mL of strain 4. While the treatment with strains 2, 3, 5, 7, and 8 (100 µg/mL) produced a significant decrease in Vero cell number percentage, P > 0.01 (cell number less than 50% with 1, 5, and 7). The treatment with strain 6 has no significant effect. B: There was a significant decrease in HeLa cell number percentage, P > 0.01 after treatment with 100 µg/mL of strain 1, 2, 3, 4, 5, 7, and 8. (cell number less than 50% except for strains 4 and 8). The treatment with strain 6 has no significant effect. C: There was a significant decrease in Hep G2 cell number percentage P > 0.05 after treatment with 100 ¼g/mL of strain 1. While the treatment with strains 2, 3, 5, 7, and 8 (100 µg/mL) produced significant decrease in Hep G2 cell number percentage P > 0.01 (cell number less than 50%). The treatment with strains 4 and 6 had no significant effect.. 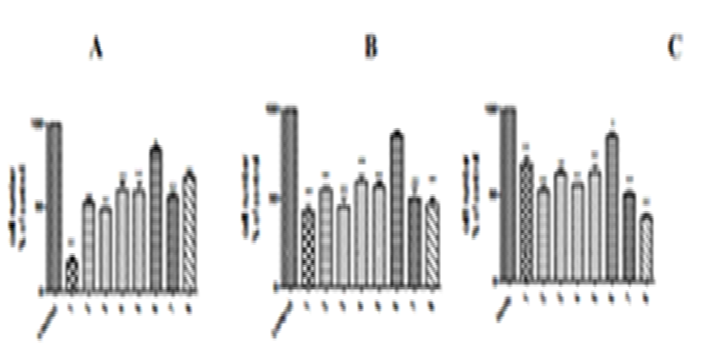 Fig. 3. A: There was a significant decrease in Vero cell number percentage, P > 0.05 after the treatment with 100 µg/mL of strain 6. While the treatment with strains 1, 2, 3, 4, 5, 7, and 8 (100 µg/mL) produced a significant decrease in Vero cell number percentage, P > 0.01 (cell number less than 50% with strains 1, 2, and 3). While strain 1 produced 70% decrease. B: There was a significant decrease in HeLa cell number percentage, P > 0.01 after the treatment with 10 µg/mL of 1, 2, 3, 4, 5, 7, and 8. (cell number less than 50% except 4 and 8). The treatment with strain 6 had no significant effect. C: There was a significant decrease in Hep G2 cell number percentage, P > 0.05 after treatment with 100 µg/mL of strain 6. While the treatment with strains 1, 2, 3, 5, 7 and 8 (10 0 µg/mL) produced a significant decrease in Hep G2 cell number percentage, P > 0.01 (cell number less than 50% with strain 8). The treatment with strains 4 and 6 had no significant effect.
Fig. 3. A: There was a significant decrease in Vero cell number percentage, P > 0.05 after the treatment with 100 µg/mL of strain 6. While the treatment with strains 1, 2, 3, 4, 5, 7, and 8 (100 µg/mL) produced a significant decrease in Vero cell number percentage, P > 0.01 (cell number less than 50% with strains 1, 2, and 3). While strain 1 produced 70% decrease. B: There was a significant decrease in HeLa cell number percentage, P > 0.01 after the treatment with 10 µg/mL of 1, 2, 3, 4, 5, 7, and 8. (cell number less than 50% except 4 and 8). The treatment with strain 6 had no significant effect. C: There was a significant decrease in Hep G2 cell number percentage, P > 0.05 after treatment with 100 µg/mL of strain 6. While the treatment with strains 1, 2, 3, 5, 7 and 8 (10 0 µg/mL) produced a significant decrease in Hep G2 cell number percentage, P > 0.01 (cell number less than 50% with strain 8). The treatment with strains 4 and 6 had no significant effect.Table (2):
A: Cytotoxicity assays CCK-8 of Hep G2 cells. B: Cytotoxicity assays CCK-8 of HeLa cells. C: Cytotoxicity assays CCK-8 of Vero.
| Dose(24Hrs) | Control(C) | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | |
|---|---|---|---|---|---|---|---|---|---|---|
| A: Hep G2 cell line (CCK-8) | ||||||||||
| 100 µg/ml | Mean | 100.0 | 51.19 | 38.33 | 36.47 | 64.32 | 41.73 | 61.90 | 34.13 | 38.82 |
| SE | 0.0 | 14.36 | 7.354 | 5.027 | 14.79 | 11.47 | 20.06 | 9.781 | 6.787 | |
| P value | P < 0.05 | P < 0.01 | P < 0.01 | P > 0.05 | P < 0.01 | P > 0.05 | P < 0.01 | P < 0.01 | ||
| B: HeLa cell line (CCK-8) | ||||||||||
| 100 µg/ml | Dose(24Hrs) | Control(C) | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
| Mean | 100.0 | 46.66 | 41.75 | 44.97 | 66.59 | 41.94 | 92.59 | 40.75 | 56.36 | |
| SE | 0.0 | 1.806 | 3.067 | 1.460 | 0.9774 | 0.2391 | 5.242 | 6.004 | 9.014 | |
| P value | P <0.01 | P <0.01 | P < 0.01 | P < 0.01 | P < 0.01 | P > 0.05 | P < 0.01 | P <0.01 | ||
| C:Vero cell line (CCK-8) | ||||||||||
| 100 µg/ml | 2 | 3 | 4 | 5 | 6 | 7 | 8 | |||
| Mean | 100.0 | 48.66 | 72.73 | 60.35 | 80.97 | 54.66 | 100 | 53.97 | 67.38 | |
| SE | 0.0 | 4.688 | 4.888 | 2.529 | 0.9511 | 2.281 | 0.0 | 1.985 | 1.400 | |
| P value | P<0.01 | P<0.01 | P<0.01 | P<0.05 | P<0.01 | P<0.05 | P<0.01 | P<0.01 | ||
Table (3):
A: Anti-proliferation of Vero cells. B: Anti-proliferation of HeLa cells. C: Anti-proliferation of Hep G2 cells.
| Dose(24Hrs) | Control(C) | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | |
|---|---|---|---|---|---|---|---|---|---|---|
| A: Vero cell line MTT | ||||||||||
| 100 µg/ml | Mean | 100.0 | 18.00 | 53.06 | 49.16 | 60.63 | 59.72 | 84.58 | 56.81 | 68.50 |
| SE | 0.0 | 2.423 | 1.170 | 0.8328 | 4.210 | 4.865 | 1.802 | 2.435 | 1.166 | |
| P value | P<0.01 | P<0.01 | P<0.01 | P<0.01 | P<0.01 | P<0.01 | P<0.01 | P<0.01 | ||
| B: HeLa cell line MTT | ||||||||||
| 100 µg/ml | Dose(24Hrs) | Control(C) | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
| Mean | 100.0 | 42.54 | 54.83 | 45.37 | 59.45 | 56.71 | 85.21 | 49.96 | 46.56 | |
| SE | 0.0 | 3.323 | 1.221 | 5.093 | 3.234 | 2.076 | 1.915 | 5.927 | 3.213 | |
| P value | P<0.01 | P<0.01 | P<0.01 | P<0.01 | P<0.01 | P>0.05 | P<0.01 | P<0.01 | ||
| C: Hep G2 cell line MTT | ||||||||||
| 100 µg/ml | Dose(24Hrs) | Control(C) | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
| Mean | 100.0 | 42.54 | 54.83 | 45.37 | 59.45 | 56.71 | 85.21 | 49.96 | 46.56 | |
| SE | 0.0 | 3.323 | 1.221 | 5.093 | 3.234 | 2.076 | 1.915 | 5.927 | 3.213 | |
| P value | P<0.01 | P<0.01 | P<0.01 | P<0.01 | P<0.01 | P<0.05 | P<0.01 | P<0.01 | ||
Over the last few years, a significant amount of effort has been exerted toward the development of wild-type or attenuated bacterial and viral strains that have a potential for the treatment of cancer. However, in Saudi Arabia, our knowledge regarding the presence of bacteria in indigenous soil habitats is extremely limited (Assaeedi et al., 2011). The primary objective of this study was to investigate if B. thuringiensis is an intrinsic part of the terrestrial environment of The Makkah province. Our results indicated that B. thuringiensis is naturally present in agricultural as well as urban soil but is absent from soil found at the beach. Our results are in accordance with those reported previously wherein it has been stated that the bacterium is more frequently recovered from agriculture-based soils and, to a lesser degree, from urban soils; however, in the beach soil or desert sand, the incidence of the bacterium is either very low or altogether absent (Quesada et al., 2004). This phenomenon can probably be explained by variations in the biotic and abiotic components of the different soil habitats and their potential impact on the distribution of B. thuringiensis. In support of this hypothesis, we found dead larvae of the cotton leaf worm, regarded as an entomo fauna of economic importance in Saudi Arabia, in the Makkah area (Yuichi et al., 2007). It is important to note that all samples analyzed in this project were collected from the areas with no previous history of using any B. thuringiensis based commercial product for control of insect or pests. Thus, we propose that all the soil and dead insect-derived B. thuringiensis isolates that were recovered in our study were an indigenous part of the microflora of the area that we explored. The frequency of B. thuringiensis recovery was only 4.6% among the soil population of Bacillus-like colony. This number is higher as compared to those reported for soil samples of other Asian countries (Poornima et al., 2010). Phase contrast microscopy showed that the inclusion bodies of the isolates recovered in our study were spherical in shape and often bigger than the spores themselves; this is in the agreement with other studies (Mizuki et al., 1999). None of our isolates were observed to produce a lepidopteran specific bipyramidal crystal that was reportedly observed to be produced by environmental isolates of B. thuringiensis (Poornima et al., 2010). The spherical crystalline inclusions that were commonly observed in our isolates are consistent with previous observations wherein it has been stated that such inclusions are usually associated with dipteran specific strains or non-insecticidal strains of Bt (Nadarajah et al., 2008). We used the 16S rRNA gene sequence to probe the extent of the phylogenetic relationship between various isolated strains. The results revealed that the isolates have 98% identity with B. thuringiensis. All samples were seen to produce significant alterations in the three cancer cell lines (Vero, HeLa, and Hep G2) that were used in this study. As a result of this observation, we propose that the indigenous strains of B. thuringiensis isolated in our study can be potentially classified as cancer cell-killing strains. Further molecular characterization of this protein will enable us to use it as a therapeutic agent for cancer therapy.
As a result of this observation, we propose that the indigenous strains of B. thuringiensis isolated in our study can be potentially classified as cancer cell-killing strains. Further molecular characterization of this protein will enable us to use it as a therapeutic agent for cancer therapy.
ACKNOWLEDGMENTS
This project was fully funded by King Abdulaziz City for Science and Technology General Directorate of Research Grants Program Project No: TK – 11- 0574. Kingdom of Saudi Arabia.
- Abulreesh, A., Osman, G.E.H., Assaeedi, A.S.A., Characterization of insecticidal genes of Bacillus thuringiensis strains isolated from arid environments. Ind J Microbiol.. 2012; 3:500–3.
- Alley, M.C., Scudiero, D.A., Monks, A., Hursey Czerwinski,.M.J., Fine, D.L., Abbott, B.J., Mayo, J.G., Shoemaker, R.H., and Boyd, M.R. Feasibility of drug screening with panels of human tumor cell lines using a microculture tetrazolium assay. Cancer Research., 1988; 48:3, 589-601.
- Assaeedi, A.S.A., Osman, G.E.H., Abulreesh, H.H., The occurrence and insecticidal activity of Bacillus thuringiensis in the arid environments. AJCS., 2011; 5:1185–1190.
- de Maargd, R.A., Bravo, A., Berry, C., Crickmore, N., Schnepf, H.E. Structure, diversity, and evolution of protein toxins from spore-forming entomopathogenic bacteria. Annu Rev Genet., 2003; 37:409–433.
- Bradford, M.M., A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem., 1976; 72:248-254.
- EL-Ghareeb, D.K., Osman, G.H., El baz, A.F. Isolation, cloning, and overexpression of vip3Aa gene isolated from a local Bacillus thuringiensis. Biocontrol Science & Technology., 2012; 22(1):11–21.
- El-Menofy, W.H., Osman, G.H., Assaeedi, A., Salama, M.S., A Novel Recombinant Baculovirus overexpressing a Bacillus thuringiensis Cry1Ab toxin enhances insecticidal activity. Biol Proced Online., 2014; 1:16″7.
- Yuichi Abe, Hiroyasu Shimada., Sakae Kitada, Raft-targeting and Oligomerization of Parasporin-2, a Bacillus thuringiensis Crystal Protein with Anti-Tumour Activity., J of Biochem., 2007; 143 ( 2) : 269-275.
- Ishiyama, M., Tominaga, H., Shiga, M., Sasamoto, K., Ohkura, Y., Ueno K. A combined assay of cell viability and in vitro cytotoxicity with a highly water-soluble tetrazolium salt, neutral red and crystal violet. Biol Pharm Bull., 1996;11: 1518–1520.
- Ito, A., Sasaguri, Y., Kitada, S., Kusaka, Y., Kuwano, K., Masutomi, K., Mizuki, E., Akao, T., Ohba, M.A., Bacillus thuringiensis crystal protein with selective cytocidal action to human cells.. J Biol Chem., 2004; 279:21282–6.
- Katayama, H., Yokota, H., Akao, T., Nakamura, O., Ohba M, Mekada E, Mizuki, E. Parasporin–1, a novel cytotoxic protein to human cells from non-insecticidal parasporal inclusions of Bacillus thuringiensis. J Biochem., 2005; 137:17–25.
- Kitada, S., Abe, Y., Shimada, H., Kusaka, Y., Matsuo, Y., Katayama, H., Okumura, S., Akao, T., Mizuki, E., Kuge, O., Sasaguri, Y., Ohba, M., Ito, A. Cytocidal actions of parasporin–2, an anti-tumor crystal toxin from Bacillus thuringiensis. J Biol Chem., 2006; 281:26350–60.
- Kuroda, S., Begum, A., Saga, M., Hirao, A., Mizuki, E., Sakai, H., Hayakawa. Parasporin 1Ac2, A novel cytotoxic crystal protein isolated from Bacillus thuringiensis B0462 strain. Curr Microbiol., 2013; 66:475–80.
- Meky, F. A., Hardie, L. J., Evans, S. W. Wild, C. P. Deoxynivalenol induce dimmunomodulation of human lymphocyte proliferation and cytokine production. Food Chemical Toxicology., 2001; 39: 827-836.
- Manes, S., del Real, G., Martinez, A.C. Pathogens: raft hijackers. Nat Rev Immunol., 2003; 3:557–68.
- Mizuki, E., Ichimatsu, T., Hwang, S.H., Park, Y.S., Saitoh, H., HiguchiK, Ohba, M. Ubiquity of Bacillus thuringiensis onphylloplanes of arboreous and herbaceous plants of Japan. J Appl Microbiol., 1999; 86:979–84.
- Mizuki, E., Park ,Y.S., Saitoh, H., Yamashita, S., Akao, T., Higuchi, K., Ohba, M. Parasporin, a human leukemic cell-recognizing parasporal protein of Bacillus thuringiensis. Clin Diagn Lab Immunol., 2000; 7:625–34.
- Mohamed, E.A.H., Abe, M., Ghanem, K.M., Abdel-Fattah, Y.R., Nakagawa, Y., El-Helow, E.R., Diversity of Bacillus genotypes in soil samples from El-Omayed biosphere reserve in Egypt. J Culture Collec., 2006; 5:78–84.
- Nadarajah, V.D., Ting, D., Chan, K.K., Mohamed, S.M., Kanakeswary, K., Lee, H.L. Selective cytotoxic activity against leukemic cell lines from mosquitocidal Bacillus thuringiensis parasporal inclusions. Southeast Asian J Trop Med Publ Health., 2008: 39:235–245.
- Okassov, A., Nersesyan, A., Kitada, S., Ilin, A. Parasporins as new natural anticancer agents: a review. JBUON., 2015; 20(1):5–16.
- Ohba, M., Aizawa, K. Insect toxicity of Bacillus thuringiensis isolated from soils of Japan. J Invertebr Pathol., 1986; 47:12–20.
- Osman, G., Already, R., Assaeedi, A., Organji, S., El-Ghareeb, D., Abulreesh, H., Althubiani, A.S. Bioinsecticide Bacillus thuringiensis a comprehensive Review. Egyptian Journal of Biological Pest Control. 2015; 25 (1):271–88.
- Osman, G., Mostafa, S., Mohamed, S.H., Antagonistic Activities of some streptomycete isolates against some phytopathogens. Pak J Biotecnol., 2007; 4(1–2):65–71.
- Poornima, K., Saranya ,V., Abirami, P., Binuramesh, C., Suguna, P., Selvanayagam, P., Rajaiah, S. Phenotypic and genotypic characterization of Bt LDC 391 strain that produce cytocidal proteins against human cancer cells. Biomedical Bioinformatics., 2012 8:461–465.
- Quesada-Morage, E., García-Tóvar, E., Valverde-García, P., Santiago-Álvarez, C. Isolation, geographical diversity and insecticidal activity of Bacillus thuringiensis from soils in Spain. Microbiol Res., 2004; 159:59–71.
- Saitoh, H., Okumura, S., Ishikawa, T., Akao, T., Mizuki, E., Ohba, M. Investigation of a novel Bacillus thuringiensis gene encoding a parasporal protein, parasporin–4, that preferentially kills human cancer cells. Biosci Biotechnol Biochem., 2006; 70:2935–71.
- Sanger, F., Nicklen, S., Coulson, A.R. Biochemistry DNA Sequencing with chain terminating inhibitors (DNA Polymerase/nucleotide sequence bacteriophage 4X174), Proc Natl Acad Sci., 1977; 74:5463-67.
- Schnepf, E., Crickmore, N., Van, Rie. J., Lereclus, D., Baum, J., Feitelson, J., Zeigler, D.R., Dean, D.H. Bacillus thuringiensis and its pesticidal crystal proteins. Microbiol Mol Biol Rev., 1998; 62:775–806
- Travers, R.S., Marin, P.A.W., Reichelderfer, C.F. Selective isolation of soil Bacillus spp. Appl Environ Microbiol., 1987; 53:1263–6.
- Thomas, W.E., Ellar, D.J.J. Bacillus thuringiensis var israelensis crystal deltaendotoxin: effects on insect and mammalian cells in vitro and in vivo. , J Cell Sci., 1983; 60:181-197.
- Waheed, A.A., Shimada, Y., Heijnen, H.F., Nakamura, M., Inomata, M., Hayashi, M., Iwashita, S., Slot, J.W., Ohno-Iwashita, Y. Selective binding of perfringo lysin O derivative to cholesterol-rich membrane microdomains(rafts). Proc Natl Acad Sci., 2001; 98:4926–31.
- Yamashita, S., Katayama, H., Saitoh, H., Akao, T., Park, Y.S., Mizuki, E., Ohba, M., Ito, A. Typical three-domain Cry proteins of Bacillus thuringiensis strain A1462 exhibit cytocidal activity on limited human cancer cells. J Biochem., 2005; 138:663–72.
- Yasutake, K., Uemori, A., Binh, N.D., Mizuki, E., Ohba, M. Identification parasporin genes in Vietnamese isolates of B. thuringiensis. Z Naturforsch., 2008; 63c:139–43.
© The Author(s) 2017. Open Access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License which permits unrestricted use, sharing, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.