


ISSN: 0973-7510
E-ISSN: 2581-690X
Microbial sources for phospholipase A1 (PLA1) are economic and industry relevant for degumming of oils and also their product lyso-phospholipids have been widely used as emulsifying agent. Numerous PLAs1 have been reported, but still the few enzymes have got position in the commercial sector. Due to enormous demand of PLA1 in the industrial sector, the present study was carried out to optimize PLA1 production using cheaper agro-industrial waste like defatted rice bran. For this, defatted rice bran was used in solid state fermentation for the production of PLA1.One-factor at a time approach was used to obtain maximum production of 51.5 U/gm which is 2.15 folds more than un-optimized medium. The optimized medium components were (pH 7): defatted rice bran (5gm), glucose (1% w/v), peptone (1% w/v) and olive oil (0.5 % v/v) with moisture content of 1:1.5 and after 48h of incubation at 37°C. This approach will provide the cleaner solution for degumming of oil as compare to the acid degumming and also help to reduce the stress of the environment by utilizing waste. This is the first report where in Bacillus subtilis subsp. inaquosorum was employed for PLA1 production via solid state fermentation.
Submerged fermentation, Solid-State Fermentation, Optimization, Classical approach, PLA1, Bacillis subtilis subsp. inaquosorum
Phospholipases are one of key enzymes in oil refinery industry and play a crucial role in degumming of edible oils. Physical refining using water removes only hydratable phospholipids so there is great demand for the environment friendly technique that can also remove the non-hydratable phospholipids1 from edible oils. Various chemical processes are also used by the industries to remove phospholipids that includes acid degumming but enzymatic degumming using phospholipases is more advantageous and effective2 because these enzymes can easily convert non-hydratable phospholipids to the hydratable phospholipids and can be further eliminated by centrifugation3.
Soil is a major reservoir for a variety of microorganisms4-6 and can be used for exploiting isolates possessing commercially important enzymes like PLA1. Lyso-phospholipids, the product formed by action of PLA1 on phospholipids are also in great demand as emulsifying agent, cosmetic agent as well as drug delivery agent. Various PLA1 has been purified from various mammalian systems but to obtain high titers of PLA1 microbial sources are still in demand7.
Phospholipases are basically classified into four groups depending on the site of ester bond attacked i.e. phospholipase A (PLA), phospholipase B (PLB), phospholipase C(PLC) and phospholipase D (PLD)8. Phospholipases A is further categorized into Phospholipases A1 (PLA1) and Phospholipases A2 (PLA2). Numerous PLA1have been reported from a variety of microorganism that includes PLA1 from Serritia liquifaciens9-11, Fusarium sp.12, Aspergillus oryzae13-15, Streptomyces16. Out of which only a few enzymes are available commercially viz. PLA1 from Novozymes (Fusarium PLA1) and Genecor (Streptomyces PLA1).
Fermentation process is the way out for producing high volume of enzymes. Solid state fermentation is an efficient method for enzyme production with less cost and reduced risk of contamination17-19. For solid state fermentation, agro-industrial waste can be used as a substrate. Keeping that in mind, the present study aims for production of extracellular PLA1 by using defatted rice bran as fermentation substrate.
Isolation of microorganism from soil samples
Twenty soil samples were collected from various sites like Dhaba, Mandir, Temples and Motor -market in different regions of Chandigarh, Mohali and Shimla. One gm of soil was suspended in 10 ml followed by serial dilution. Isolates were plated on tributyrin (0.5 % v/v) agar plate and incubated at 37ºC for 24h. Isolates producing clear zone indicate lipase positive culture.
Qualitative assay for PLA1
Lipase positive cultures were patched on egg-yolk (5% v/v) agar plate and incubated at 37°C for 72h. Clear zone indicates phospholipase A positive cultures20. Hydrolysis capacity (HC) ratio was measured by dividing the clearing zone diameter by colony growth diameter21.The glycerol stock of phospholipase A (PLA) positive cultures were preserved at -80°C. PLA positive cultures were selected for further studies. HC was calculated after 24h.
Gram staining characteristic
A loopful of overnight grown PLA positive culture in distilled water was put on a clean glass slide and a uniform smear was prepared. The smear was heat fixed by gently heating over the burner flame. After heat fixation the smear was stained with crystal violet complex for about one minute. The slide was gently washed under tap water. Next, smear was flooded with Gram’s iodine for about one minute and then slide was again washed gently with water. Decolourization of the smear was done by covering the smear with de-staining solution for 15-20 sec. Slide was again washed with water. Lastly, the smear was stained with safranin for about 40 sec followed by washing with water. Slide was blot dried and further examined under microscope (Quasmo, India) under 100X magnification.
PLA1 production by selected isolates via submerged fermentation
PLA positive cultures were first subjected to submerged fermentation using Nutrient Broth (NB) supplemented with olive oil (0.5% v/v). The emulsion was formed with gum- acacia (1% w/v) in mixer grinder. An inoculum was prepared in NB until O.D600 reached 0.8 and was added (1% v/v) to the production medium followed by incubation at 37°C in orbital shaker (Remi orbital shaker, India) with agitation speed 160 rpm for 24h. The medium was centrifuged at 10,000 rpm for 10minutes (Sigma Laborzentrifugen, Germany) and PLA1 activity assay was carried out with the supernatant.
Quantitative assay for PLA1 activity
The quantitation of PLA1 activity was done by using ‘Enzchek® Phospholipase A1 assay kit’ (Molecular Probes Inc. USA) that uses PLA1 dye-labeled glycerophosphoethanolamines with dye labeled acyl chain at the sn-1 position and dinitrophenyl quencher-modified head group. The standard curve for assay was prepared using lecitase provided in the kit.
Substrate-liposome mix was prepared by adding 50µl lipid-mix {30µl of 10mM of DOPC (Dioleoylphosphatidylcholine), 30µl of 10mM DOPG (Dioleoylphosphatidylglycerol) and 30µl of 2mM PLA1 substrate} slowly and steadily to the 5 ml of reaction buffer (1X) placed on a magnetic stirrer.The reaction was started by adding 50µL of substrate-liposome mix to each well of 96-well plate already containing controls and samples. This was followed by incubation at 25°C for 30 minutes in dark and fluorescence was measured with excitation at 460nm and emission at 515 nm. One Unit (U) of the enzyme corresponds to one Lecitase® Ultra unit as provided in Enzchek® Phospholipase A1 assay kit.
Identification by 16S rRNA gene sequencing
Two isolates with maximum PLA1 activity were identified using 16S rRNA gene sequencing at IMTECH, Chandigarh, India. Genomic DNA was extracted from pure cultures using HiPurATM Bacteria Genomic DNA Miniprep Purification Spin Kit (Hi-Media, India) according to the manufacturer’s instructions. The amplification of 16S rRNA gene was achieved using the following primers: 27f (5’-AGA GTT TGA TCC TGG CTC AG-3’) and 1492r (3’-ACG GCT ACC TTG TTA CGA CTT-5’). Four sequencing primers and Big-Dye Terminator Cycle Sequencing Kit (Applied Biosystems, CA, USA) were used for the sequencing of the purified 16S rRNA gene.
Sequence assembly and phylogenetic analysis
DNA sequence assembling software SEQUENCHER™ 4.10.1 (Gene Codes Corporation, MI, USA) was used to assemble and analyse the sequence data obtained. In order to find out the similar sequences, the data was used to BLAST as query in nucleotide database of NCBI (http://www.ncbi.nlm.nib.gov/). Phylogenetic tree was constructed using Clustal W software by aligning all related and acquired sequences. Neighbour-joining method was used to measure the evolutionary distances and operated in MEGA 6 and the Kimura 2 parameter models22.
PLA1 production by solid state fermentation
The identified and selected isolate with maximum extracellular PLA1 activity in submerged conditions were subjected to solid state fermentation using defatted rice bran as substrate.
The olive oil emulsion (0.5% v/v) was formed in mixer grinder using 1% (w/v) gum acacia as emulsifying agent. An inoculum (1.5ml) was obtained in NB until O.D600 reaches 0.8 and added to autoclaved defatted rice bran (5gm) with 6ml of olive oil emulsion maintaining surface to moisture content ratio of 1:1.5. The flasks were incubated at 37°C under static conditions for 48h.The content of the flask were suspended in 100ml of distilled water and kept at shaking 160rpm for 1h. Then centrifugation was carried out at 10,000rpm for 10minutes and supernatant was used for PLA1 activity assay.
Classical approach for optimization in solid state fermentation
Different parameters like incubation time, incubation temperature, moisture conditions, oil sources, nitrogen and carbon sources were taken in account to optimize the culture conditions for maximum production of PLA1 by solid state fermentation.
Incubation time
For optimization of incubation time, medium was prepared consisting of 5g defatted rice bran with 6 ml olive oil emulsion (0.5%, pH 7). The stirring of olive oil with water was carried out in a mixer grinder with 1% (w/v) gum acacia to prepare olive oil emulsion. The medium was sterilized and inoculum (1.5 ml) was added and incubated at 37ºC for 24 to 120 h.
Incubation temperature
The optimum temperature was determined by incubating the same medium at different temperatures (25-45 ºC) for 48h.
Moisture content
The moisture content in the medium was optimized by incubating 5gm defatted rice bran and inoculums (1.5 ml) with following substrate to moisture ratio (rice bran : olive emulsion + inoculums): 1:1, 1:1.5; 1:2; 1:2.5; 1:3; 1:3.5 (pH 7) and medium was incubated at 37ºC, for 48h.
Oil source
Different oil sources (0.5 % v/v) were used as inducer for PLA1 production like sunflower oil, olive oil, soy-bean and groundnut oil. The moisture content of the medium was maintained at 1:1.5 by using 6ml of oil emulsion with 1.5ml of inoculums in 5gm defatted rice bran and incubated at 37ºC for 48h.
Carbon source
The carbon source in the medium was optimized by taking different carbon sources (1% w/v) like glucose, sucrose, fructose, xylose, maltose in the same medium and incubated at 37ºC for 48h.
Nitrogen source
The nitrogen source in the medium was optimized using 1% (w/v) of malt extract, yeast extract, peptone, urea and soybean meal in the medium and was incubated at 37ºC for 48h under static conditions.
Isolation of cultures from soil samples
In total, 82 lipase positive isolates were screened on the basis of degradation of tributyrin in lipase plate assay. Out of 82, only 31 isolates were found to have PLA activity on the basis of formation clear zone on egg-yolk agar plate. The Gram staining and HC on egg-yolk agar plate of the selected 31 isolates is displayed in Table 1.
Table (1):
Gram Character, Hydrolysis Capacity
No |
Isolate number |
Gram Staining |
HC |
|---|---|---|---|
1 |
S2-1 |
Isolated, thin, Gram positive rods |
0.11 |
2 |
S3-1 |
Isolated, Gram positive rods |
1.55 |
3 |
S3-2 |
Isolated, Gram positive rods |
1.37 |
4 |
S6-3 |
Very small, Gram negative rods |
1.36 |
5 |
S7-2 |
Small, Gram positive rods |
1.60 |
6 |
S7-5 |
Very small, isolated Gram negative rods |
1.72 |
7 |
S7-6 |
Isolated, Gram negative rods |
1.25 |
8 |
S8-1 |
Gram negative rods |
1.50 |
9 |
S8-3 |
Gram positive rods in chains |
3.00 |
10 |
S9-1 |
Isolated, Gram positive thick rods |
1.40 |
11 |
S9-2 |
Small, isolated, Gram negative rods |
1.25 |
12 |
S10-2 |
Isolated, Gram positive rods |
1.44 |
13 |
S10-7 |
Isolated, Gram positive rods |
1.20 |
14 |
S11-4 |
Very small, Gram negative rods |
1.18 |
15 |
S12-2 |
Small, Gram positive rods |
1.75 |
16 |
S12-3 |
Gram positive rods in chains |
1.44 |
17 |
S13-1 |
Gram negative rods |
1.37 |
18 |
S14-1 |
Gram negative, cocco-bacilli, |
1.90 |
19 |
S14-2 |
Gram positive cocci |
1.28 |
20 |
S14-3 |
Very small, Gram negative rods |
1.44 |
21 |
S15-1 |
Gram negative cocci |
1.87 |
22 |
S15-3 |
Small, thin, Gram negative rods |
1.76 |
23 |
S16-3 |
Gram negative rods |
1.50 |
24 |
S16-4 |
Very small, Gram positive cocci |
1.75 |
25 |
S16-5 |
Gram negative cocci |
1.62 |
26 |
S17-4 |
Gram negative cocci |
1.05 |
27 |
S17-5 |
Gram positive cocci |
1.60 |
28 |
S18-1 |
Gram negative thin small rods |
1.50 |
29 |
S20-2 |
Gram positive rods, isolated |
1.36 |
30 |
S20-3 |
Gram positive rods, in chains |
1.38 |
31 |
S20-4 |
Gram positive rods, in chains |
1.50 |
PLA1 production by submerged fermentation
The PLA positive isolates were subjected to submerged fermentation. The supernatant activity of 31 isolates (U/ml) is displayed in Table 2.
Table (2):
PLA1 Activity (Submerged Conditions)
S.No. |
Isolate Number |
PLA1 activity (U/ml) |
|---|---|---|
1 |
S2-1 |
0.614 |
2 |
S3-1 |
0.133 |
3 |
S3-2 |
0.005 |
4 |
S6-3 |
0.125 |
5 |
S7-2 |
0.014 |
6 |
S7-5 |
0.472 |
7 |
S7-6 |
0.087 |
8 |
S8-1 |
0.394 |
9 |
S8-3 |
1.250 |
10 |
S9-1 |
0.839 |
11 |
S9-2 |
0.064 |
12 |
S10-2 |
0.013 |
13 |
S10-7 |
0.023 |
14 |
S11-4 |
0.019 |
15 |
S12-2 |
0.021 |
16 |
S12-3 |
0.241 |
17 |
S13-1 |
0.404 |
18 |
S14-1 |
1.170 |
19 |
S14-2 |
0.071 |
20 |
S14-3 |
0.513 |
21 |
S15-1 |
0.016 |
22 |
S15-3 |
0.263 |
23 |
S16-3 |
0.271 |
24 |
S16-4 |
0.910 |
25 |
S16-5 |
0.097 |
26 |
S17-4 |
0.018 |
27 |
S17-5 |
0.078 |
28 |
S18-1 |
0.315 |
29 |
S20-2 |
0.043 |
30 |
S20-3 |
0.228 |
31 |
S20-4 |
0.195 |
Identification of bacteria by 16S rRNA sequencing
Two isolates with maximum PLA1 activity S8-3 and S14-1 were selected for identification. The 16S rRNA gene sequence of the S8-3 displayed 99 % identity with Bacillus subtilis subsp. inaquosorum KCTC 13429 and S14-1 strain showed 99.78 % identity with Acinetobacter radioresistens DSM 6976T. The S14-1 was not used for further studies due to its pathogenic nature. Nucleotide sequence obtained after sequencing of S8-3 was submitted in GENBANK under Accession no. KY088040 with strain name RG1. The Phylogenetic tree of both isolates was constructed by Neighbor joining method (Fig. 1) on the basis of sequence analysis.
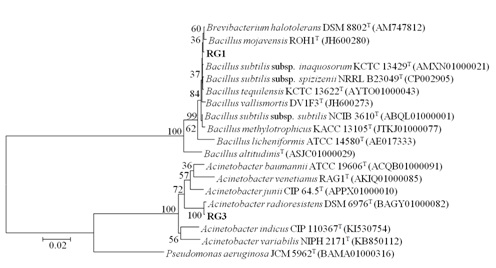
Fig. 1. Phylogenetic tree representing evolutionary relationship of RG1 and RG3: Neighbor-Joining method was used to infer evolutionary history. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1000 replicates) is shown next to the branches. The Kimura 2-parameter method was used to compute evolutionary distances and is in the units of the number of base substitutions per site. Evolutionary anal-yses were carried out using MEGA6. Pseudomonas aeruginosa JCM 5962T (BAMA01000316) was taken as an out-group. NCBI Genbank accession numbers of 16S rRNA gene sequences are shown in parenthesis
Optimization of the cultural conditions
Bacillus subtilis subsp. inaquosorum RG1 was subjected to solid state fermentation for PLA1 production. The optimization was carried out by classical approach by taking different factors in account including incubation temperature, incubation time, moisture content, different oils, different nitrogen and carbon sources (Figure 2 and Figure 3). Maximum PLA1 activity of 51.55 U/gm was obtained after the incubation of 48h at 37°C with moisture content of 1: 1.5 using glucose and peptone as carbon and nitrogen source respectively with 0.5% olive oil as an inducer.
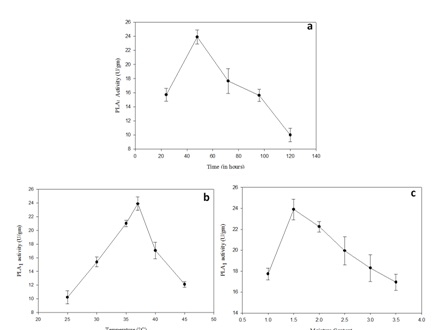
Fig. 2. Effect of incubation conditions on PLA1 production by Bacillus subtilis subsp. inaquosorum (a) incubation time (24h-120h) (b) incubation temperature (25-45˚C) (c) moisture con-tent (1:1-1:3.5) via solid state fermentation
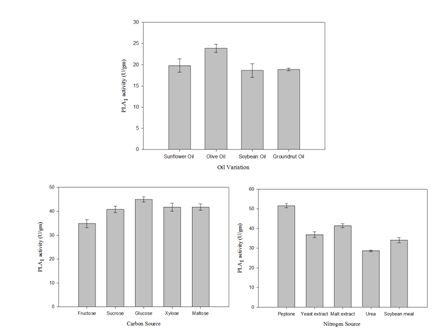
Fig. 3. The effect of medium composition on PLA1 production (a) oil variation (b) carbon sources (d) nitrogen sources
PLA1 production (23.2 U/gm) was observed after incubation of 48h and after that there is a decrease in PLA1 that may be due to decrease in viable number of cells23 and depletion of nutrient availability24.
Substrate to moisture ratio of 1:1.5 was found to be more suitable for PLA1 production. The moisture content ratio plays a crucial role in the growth of microorganism ultimately affecting enzyme productions by solid state fermentation25.
Temperature is a crucial factor for optimum enzymatic activity and overall metabolism26. In our study, optimum PLA1 production was observed at 37°C. Our results are in accordance with previous reports wherein 37°C was the best temperature for maximum PLA1 production in Pseudomonas gessardii and Trichosporon sp.27.
In addition to physical factors, medium composition also affects the enzyme production as it play an important role in overall growth of microorganisms.
Various carbon and nitrogen source and different oils have different effects on growth of microorganisms and finally influenced enzyme production. Thus, these parameters need to be optimized for better production yields of the desired enzyme 24.
In our study, 0.5% of olive oil is observed to be the best inducer for PLA1 production. Similar results were observed for lipase production from Bacillus sp.28,29. Among the carbon and nitrogen sources used, glucose and peptone were found to be the best carbon and nitrogen source respectively. In contrast, xylose and ammonium sulphate were found as good carbon and nitrogen source for PLA1 in Serriatia sp.11.
The use of PLA1 has been restricted owing to its low stability and less availability. In addition, crystallographic and structural data for most of the PLAs1 have not been obtained yet. So there is an increasing trend to isolate new microorganisms for PLA1 production which can provide a platform to further improve these microbes to make them competent enough to be exploited at industrial scale.
In the present study, a PLA1 producing microorganism was isolated from soil and identified as Bacillus subtilis subsp. inaquosorum. Maximum PLA1 production of 51.55 U/gm was observed which is 2.15 folds more than un-optimized medium via solid state fermentation. This is the first report wherein PLA1was produced using one factor at a time approach. Moreover, the present study also provided a clean approach to successfully produce high titers of enzyme and will reduce burden of the environment by utilizing waste as substrate.
ACKNOWLEDGMENTS
We would like to acknowledge Dr G.S. Prasad (IMTECH, Chandigarh) for identification of isolate and Dr. Deepak Sharma (IMTECH, Chandigarh) for their assistance in work.
- Jiang, X., Chang, M., Wang, X., Jin, Q., Wang, X. Application of phospholipase A1 and phospholipase C in degumming process of different kinds of crude oil. Process. Biochem., 2015; 50:432-437.
- Yu, D., Jiang, L., Li, Z., Shi, J., Xue, J., Kakuda, Y. Immobilization of Phospholipase A1 and its application in soybean oil degumming. J.Am.Oil.Chem., 2012; 89:649–656.
- Zhan, J., Jiang, S., Pan, I., Zhang, Y. Purification, characterization and application of a cold adapted phospholipase A1 from Bacillus cereus sp. AF-1. Biotechnol.Biotechnol.Equip., 2013; 27(4): 3972-3976.
- Baumgardner, D.J. Soil-related bacterial and fungal infections. J.Am.Board.Fam.Med., 2012; 25:734-744.
- Berg, G., Smalla, K. Plant species and soil type cooperatively shape the structure and function of microbial communities in the rhizosphere. FEMS.Microbiol.Ecol., 2009; 68:1-13.
- Strawn, D.G., Bohn, H.L., O’Connor, G.A.: Soil chemistry,4th edn. Wiley-Blackwell, 2015.
- Song, J.K., Kim, M.K., Rhee, J.S. Cloning and expression of the gene encoding phospholipase A1 from Serratia sp. MK1 in Escherichia coli. J.Biotechnol., 1999; 72: 103–114.
- Vance, J.E., Vance, D.: Biochemistry of lipids, lipoproteins and membranes, 4th edn. Elsevier Science, 2002.
- Givskov, M., Olsen, L., Molin, S. Cloning and expression in Escherichia coli of the gene for extracellular phospholipase A1 from Serratia liquefaciens.J.Bacteriol.,1998;170: 5855-5862.
- Yan, J., Zhang, L., Gu, Z., Ding, Z., Shi, G.Cloning, expression of phospholipase A1 from Serratia liquefaciens and auto-induction fermentation by lactose. J.Bacteriol., 2013; 29:853.
- Kim, M.K., Kim, J.K., Rhee, J.S. Isolation of a phospholipase A1 producing microorganism. J.Ind.Microbiol.Biotechnol., 1996; 16:171-174.
- Clausen, K. Enzymatic oil-degumming by a novel microbial phospholipase. Eur.J.Lipis.Sci.Technol., 2001; 103:333-340.
- Birch, M., Robson, G., Law, D., Denning,D.W. Evidence of multiple extracellular phospholipase activities of Aspergillus fumigatus. Infect.Immun., 1996; 64: 751-755.
- Watanabe, I., Koishi, R., Yao, Y., Tsuji, T., Serizawa, N. Molecular cloning and expression of the gene encoding a phospholipase A1 from Aspergillus oryzae.Biosci.Biotechnol.Biochem.,1999; 63:820–826.
- Shiba, Y., Ono, C., Fukui, F., Watanabe, I., Serizawa, N., Gomi, K., Yoshikawa, H. High-level secretory production of phospholipase A1 by Saccharomyces cerevisiae and Aspergillus oryzae. Biosci.Biotechnol.Biochem., 2001; 65:94-101.
- Taghrid, S.I., Peter, J.C. Phospholipase A in Gram-negative bacteria and its role in pathogenesis. Microbiology., 2006; 152:1263-1274.
- Pandey, A. Solid-state fermentation. Biocem.Eng.J., 2003; 13:81-84.
- Kamanth, P.V., Dawarakanath, B.S., Chaudhary, A., Janakiraman, S. Optimization of culture conditions for maximum lovastatin production by Aspergillus terrus (KM017963) under solid state fermentation. HAYATI.J.Biosci., 2015; 22:174-180.
- Mienda, B.S., Idi, A., Umar, A. Microbiological Features of Solid State Fermentation and its Applications – An overview. Res. Biotechnol., 2011; 2(6): 21-26.
- Chrisope, G.L., Fox, C.W., Marshall, R.T. Lecithin agar for detection of microbial phospholipases. Appl.Environ.Microbio., 1976; 31(5): 784-786.
- Akaracharanya, A., Taprig, T., Sitdhipol, J., Tanasupawat, S. Characterization of cellulase producing Bacillus and Paenibacillus strains from Thai soils. J.Appl.Pharm.Sci., 2014; 05:006-011.
- Tamura, J., Dudley, M.N., Kumar, S. Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol.Biol.Evol., 2007; 24:1596-1599.
- Swamy, M.K., Kashyap, S.S.N., Vijay, R., Tiwari, R., Anuradha, M. Production and optimization of extra cellular protease from Bacillus sp. isolated from soil. Intl.J.Adv.Biotech.Res., 2012; 3:564-569.
- Kusuma, M.P., Reddy, R.D.S. Optimization of polygalacturonase using isolated Bacillus subtilis C4 by submerged fermentation. J. Pharm.Res., 2014; 8(2):106-112.
- Malakar, R., Yadav, M., Malviya, S.N., Tiwari, A. Effect of media composition and culture condition on pullulanase enzyme from extremophile bacterial species. Int.J.Biomed.Adv.Res., 2012; 03: 692-699.
- Veerapagu, M., Narayanan, S.A., Ponmurugan, K., Jeya, K.R. Screening, selection, identification production and optimization of bacterial lipase from oil spilled soil. Asian.J.Pharm.Clin.Res., 2013; 3: 62–67.
- Bentubo, H.D.L., Gompertz, O.F. Effects of temperature and incubation time on the in vitro expression of proteases, phospholipases, lipases and DNases by different species of Trichosporon. Springer Plus., 2014; 3:377.
- Lee, D.W., Koh, Y.S., Kim, K.J., Kim, B.C., Choi. H.J., Kim, D.S. Isolation and characterization of a thermophilic lipase from Bacillus thermoleovorans ID-1. FEMS.Microbiol.Lett., 1999; 179: 393-400.
- Eltaweel, M.A., Rahman, R.N.Z.R.A., Salleh, A.B., Basri, M. An organic solvent-stable lipase from Bacillus sp. strain 42. Annu.Rev.Microbiol.,2005; 55: 187-192.
© The Author(s) 2017. Open Access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License which permits unrestricted use, sharing, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.