


ISSN: 0973-7510
E-ISSN: 2581-690X
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the causative agent of coronavirus (CoV) disease 2019 (COVID-19). This study compared the genome, mutations, and infectivity/transmissibility of SARS-CoV-2 with selected betacoronaviruses (beta-CoVs). This study further examined the origin, risk factors, and outbreaks caused by beta-CoVs. We searched the following databases for relevant studies: PubMed, Google Scholar, and the World Health Organization COVID-19 database. A close relationship between SARS-CoV-2 and SARS bat-like CoV RaTG13 (98.9%) was found at the amino acid level, followed by pangolin CoVs. Non-synonymous mutations occur at high frequencies in the open reading frame (ORF) 1ab, spike (S) protein, and nucleocapsid. Mutations P323L and D614G in the RNA-dependent RNA polymerase (RdRp) and S protein, respectively, occur at a high frequency globally. Mutations at position 3037 in the nonstructural protein (Nsp) 3, 14408 (RdRp), and 23403 (S) confer transmissibility to SARS-CoV-2. SARS-CoV-2 has higher infectivity and transmissibility than SARS-CoV, which shares the same receptor. Although bats are confirmed reservoirs, intermediate hosts are currently unknown. Smoking, old age, diabetes, cardiovascular diseases, and hypertension have all been associated with COVID-19. Within six months of its outbreak, COVID-19 was reported in all countries worldwide, whereas SARS was reported in 28 countries and Middle East respiratory syndrome (MERS) in 5 countries. However, the fatality rate of MERS (65%) was higher than that of COVID-19 (4.9%) and SARS (6.6%). Identifying the SARS-CoV-2 intermediate hosts will help prevent future outbreaks. Attention should be given to the pangolin CoVs. Variations in the S gene may confer transmissibility and infectivity.
Mutations, SARS-CoV-2, Pandemic, COVID-19, Coronaviruses, Respiratory Syndrome
Betacoronaviruses (beta-CoVs) have been responsible for three outbreaks that are of public health significance over the past two decades.Coronavirus (CoV) disease 2019 (COVID-19) first appeared in December 2019, and the World Health Organization (WHO) announced the name of the new epidemic on February 11, 2020. The virus responsible was originally called the 2019 novel CoV. However, its name was changed by the International Committee on Taxonomy of Viruses to severe acute respiratory syndrome CoV2 (SARS-CoV-2)1,2. SARS-CoV-2 is a beta-CoV belonging to the subgenus Sarbecovirus. Other organisms in the genus of public health concern are SARS-CoV and Middle East respiratory syndrome (MERS)-CoV1. MERS-CoV caused the MERS outbreak in 2012–2015, resulting in 1,621 cases and 584 deaths3. SARS-CoV was responsible for the SARS outbreak, spreading across 37 countries in 2002–2003 resulting in 8,000 cases and 774 fatalities3,4.Other human CoVs (HCoVs) are NL63 and 229E of the Alphacoronavirus genus and OC43 and HKU1 of the genus Gammacoronavirus1,3,4.
Online databases, including PubMed, Google Scholar, and the WHO COVID-19 database, were searched for articles containing the phrase “SARS-CoV-2” and any of the following terms: “genome”, “mutations”, “epidemiology”, “infectivity”, “Spike protein” or “host”. Additional search terms identified in the first round of screening were used to conduct subsequent searches in the databases. This included, “nCoV-2019” and “genome”, “risk factors”, “intermediate host” or ” Spike protein. The articles obtained in each search were reviewed based on their titles and abstracts for relevance. The SARS-CoV-2 articles were published in January 2020. The WHO situation reports on SARS and MERS outbreaks and a systematic review of CoVs were also included.
SARS-CoV-2 compared to selected coronaviruses
SARS-CoV-2 has approximately 29.9kb, non-segmented single-stranded positive RNA genome, enclosed in an envelope(E)5,6. At the beginning of the genome lies a short untranslated region (5′ UTR) followed by an open reading frame 1 (ORF1), which is approximately 67% of the RNA. ORF 1 is composed of Pp1a and Pp1ab, which encode 16 non-structural proteins involved in transcription and replication5,7–9. Nsp 12, also known as RNA-dependent RNA polymerase (RdRp), catalyzes RNA synthesis with Nsp 7 and Nsp 8 as co-factors7,10. At the 3′ terminus, there are four structural proteins: the nucleocapsid (N), membrane (M), spike (S), and E8,11.
CoVs use S glycoproteins to gain entry into potential host cell8,11. The S1 region of the S protein allows binding where as the S2 region enhances fusion. Thus, the S protein determines host tropism8,12. Peptide insertion (PRRA) in the S1/S2 junction ofthe S protein (position 681–684) induces a furin cleavage motif (RRAR) and is unique to SARS-CoV-213,14. M functions include nutrient transport, bud release, and the formation of envelope (E). In addition, together with the E protein, they participate in the assembly of viral particles. The N protein catalyzes RNA synthesis5,15.The SARS-CoV-2 genome also has eight accessory proteins: ORF 3a, 3b, 6, 7a, 7b, 8a, 9b, and 14, which are common to other CoVs5,16.
The genome of SARS-CoV-2 is more similar to the bat-SARS genome than the SARS-CoV and MERS-CoV genomes. For instance, SARS-CoV-2 is approximately 96.2% similar to the whole genome sequence of the bat CoV RaTG13 of Rhinolophus affinis, and 87%–89% similar to strains SL-CoVZC45 and SL-CoVZXC21 of Rhinolophus sinicus4,17. The SARS-CoV-2 genome is 85.5%-92.4% similar to that of Guangdong and Guangxipangolin SARS-related-CoVs18. SARS-CoV-2 has 50% and79% whole genome sequence similarity to MERS-CoV and SARS-CoV, respectively18–20. SARS-CoV-2 is similar to SARS-CoV at the amino acid (aa) level but has distinct dissimilarities. The ORF 8 gene in SARS-CoV-2 is not segmented in 8a and 8b, similar to SARS-CoV22. ORF 3b is 22aa-long in SARS-CoV-2 where as it is 154aa long in SARS-CoV16.SARS-CoV-2shares 40.2% similarity with mild respiratory HCoV-OC43 at the nucleotide level19.At the nucleotide level, the relatedness between SARS-CoV-2 and canine and bovine CoVs ranged from 40.8% to 41.5%. Comparisons between different genes showed that the S gene of SARS-CoV-2 has higher homology with that of bat CoVs (95.4%–97.7%) and pangolins (97.5%). The protein sequences of SARS-CoV-2 and SARS causative agent S genes derived from humans, civets, and bats have approximately 76%–78% similarity 22–24. The RdRp gene of SARS-CoV-2 is extremely similar (96%) to that of SARS-CoV.
We calculated the percent average of amino acid similarities of the whole genome, ORF 1ab, S, M, E, and N proteins of several CoVs to SARS-CoV-2. In case of a range, we used the highest value. Based on amino acid similarities of the entire genome and several regions (Table 1), a remarkably close relationship was observed between SARS-CoV-2 and bat-CoV RaTG13 (98.9%). This was followed by pangolin CoVs (96.2%), and SARS-CoV (85.2%). We also observed a distant relationship between SARS-CoV-2and MERS-CoV genomes based on aa similarities (40.4%) of the whole genome, ORF 1ab, S, E, M, and N.
Table (1):
Amino acid sequence similarity in % of selected coronaviruses to SARS-CoV-2.
| SARS-CoV | MERS-CoV | RaTG13 | Pangolins CoVs | References | |
|---|---|---|---|---|---|
| Whole genome | 76 | 48 | 95.8-98.5 | 87.1-94.1 | 14,22,58–62 |
| ORF 1ab | 86.1 | 45.4 | 98.5 | 87.1-90.3 | |
| Spike protein | 75.9 | 28.9 | 91.5-97.4 | 88.3-97.5 | |
| Envelope | 94.7 | 35.3 | 100 | 97.4-100 | |
| Membrane | 89.1 | 38.7 | 98.1-100 | 93-98.6 | |
| Nucleocapsid | 90.2 | 45.9 | 99 | 96.7 | |
| Average | 85.3 | 40.4 | 98.9 | 96.2 |
Mutations present on the SARS-CoV-2 genome
Similar to other RNA viruses, mutations in CoVs arise during every replication cycle4. Mutations in viruses can modulate virulence, transmission, receptor affinity, and host tropism among others6,25. Interestingly, in the order Nidovirales (to which Coronaviridae belongs) RNA polymerase has a proof reading mechanism.Nsp 14 in SARS-CoV has a proof reading function that increases the fidelity of RNA synthesis during exonuclease activity. It is expected to carry out the same function in SARS-CoV-2 correcting errors by RdRp26. Thousands of mutations, ranging from synonymous to non-synonymous, insertions, and deletions, have been reported in more than 50,000 genome sequences that were uploaded in the Global Initiative on Sharing all Influenza Data and the NCBI database. Approximately 80% of the reported mutations were found to be non-synonymous27.
We reviewed studies that compared 10 Chinese isolate sequences to more than 1,000 sequences that have been uploaded from around the world. Mutations have been found to occur at high frequencies in the ORF 1ab, S, and N genes,as well as ORF3a6,28,29. No mutations were detected in the Chinese isolates in December 20194. The first non-synonymous mutation to be reported was in position 28151 in ORF 8, resulting in serine and leucine (S and L) clade. Position 28151 within ORF 8 is characterized by a cytosine or uracil that codes fora serine/leucine (S/L) variation at aa 84. Amino acid 84 is not conserved among the other CoVs. The S-Clade was the original virus but it became less prevalent in course of time (approximately 30%). The prevalence of the L-Clade was approximately 70% early in the outbreak but its frequency decreased in late January 2020. It was more aggressive than the S-clade5,18,30. The SARS-CoV-2 S-clade is characterized by a mutation at position 28151. The strains isolated in North America and Europe were of the L-variant descent26,29.
The other significant mutation was located at position 14408 of the RdRp gene. It is anticipated to have appeared first in Italy on February 20, 2020, resulting in an aa change P323L.At this time, the number of individuals contracting the virus increased substantially in Europe26,31.Viruses with the P14408L mutation have a higher mutation rate (median of three point mutations) than those without the mutation, with a median of one point mutation 26. Clade G of SARS-CoV-2 is based on a mutation in the S gene at position 23403, resulting in D614G and became the dorminant circulating variant as of June 2020. Clade V is based on a variation in position 26143 which resulted in aa change G251V in ORF 3a6,29. Subclades G.1 and G.2 are the result of the following mutations: G204R and R203K in N gene,P214L in ORF1b, and Q57H in ORF3a6. Mutations 2891 and 3036 (Nsp 3), 14408 (Nsp 12), 23403 (S protein),28881G > A, 28882G > A, and 28883G > C (N) are characteristic of isolates from Europe26.Mutations 1059C>T, 18060 (Nsp 14), 17857, and 17746 (Nsp 13, helicase),and 25563G>T are the characteristics of North American isolates26,32. According to Pachetti et al. (2020)26, mutations at the position 241 in the leader sequence, 3037 (Nsp 3), 14408 (RdRp),and 23403 (S), confer transmissibility. Non-synonymous mutations 241 (leader sequence), L37F (Nsp 6), G251V, P323L, D614G (S gene), L84S (ORF 8), and synonymous mutations S75S (Nsp 4) and F105F (Nsp 3) occur in high-frequency globally6,29,33. Two deletions of three nucleotides and 24nt in the ORF 1ab and one deletion of 10nt at the 32 end of the genome were observed in sequences of SARS-CoV-2 obtained from Aichi, Japan; Wisconsin,USA; and Victoria, Australia34. Table 2 summarizes the mutations that occur in different regions of the SARS-CoV-2 genome. The majority of mutations have been reported in ORF 1ab (n=59), S (n =32), and N (n=14). Figure 1 shows the logarithm (Log10) of mutations occurring in regions with more than five mutations.
Table (2):
Mutations occurring on the SARS-CoV-2 genome.
Region |
Genomic position |
Non synonymous |
Author |
|---|---|---|---|
5’UTR |
C241T |
||
ORF 1ab |
266-21555 |
N=59 L36F, L37F, A88V, H116Q, P118L, A117T, A144V, P214L, T266I, H286Y, P287T, P308S, P 309S , P314L, P323L, G392D, S 428N , P504L, C541Y, T609I, T708I, I739V, P765S, H819Y, A876T, A1043V, A1176V, V1397I, L1599F, A1606T, I1607V, M2194T , L2235I, I2244T, G2251S, A2345 V, G2534V, D2579A, N2708S, N2894D, F2908 I , T3058I, F3071Y, S3099L, G3334S, L3606F, E3764 D, N3833K , L3691F, S4396L, W5308C, T5579I, I6074V, I 6075 T, P6083 L, F6309Y , E6565D, K6958R, D7018N |
9,21,26,29,33,34,63,64 |
Spike Protein |
21563-25384 |
N=32 Y28N, T29I, F32I, H48Y, H49, L54F, N74K, E96D, D111N, F157L, G181V, S221W, S 247 R, A348T, N354D, D 364Y, V366F, V367 F, R408, G476S, V483A, H519Q, A520S, D 614G, A930V, D936Y, A1078V, G1124V, P1143L, D1168H, E1207V, D1259H |
9,26,28,34,56,64,65 |
ORF 3a |
25393-26220 |
N=3 Q57H, G196V, I208T, G251V |
33,64,66 |
Envelope |
26245-26472 |
N=6 L37H, S50G, V52I, V58L,S68C, L73F |
6 |
Matrix |
26523-27191 |
N=3 T175M, D209H, I1208T |
29,34,66 |
ORF 6 |
27202-27387 |
||
ORF 7a |
27394-27759 |
||
ORF 7b |
27756-27887 |
||
ORF 8 |
27894-28259 |
N=3 V32L, V61L, L84S |
(34, 40) |
Nucleocapsid |
28274-29533 |
N=14 P46S, D103Y, T148I , S193I S194L, S197L, S202N , R203K, G204R, G238C, D249H, K343I, P344S, S404A |
(22,30,37,38,40,41) |
ORF 10 |
29558-29674 |
R9659H |
67 |
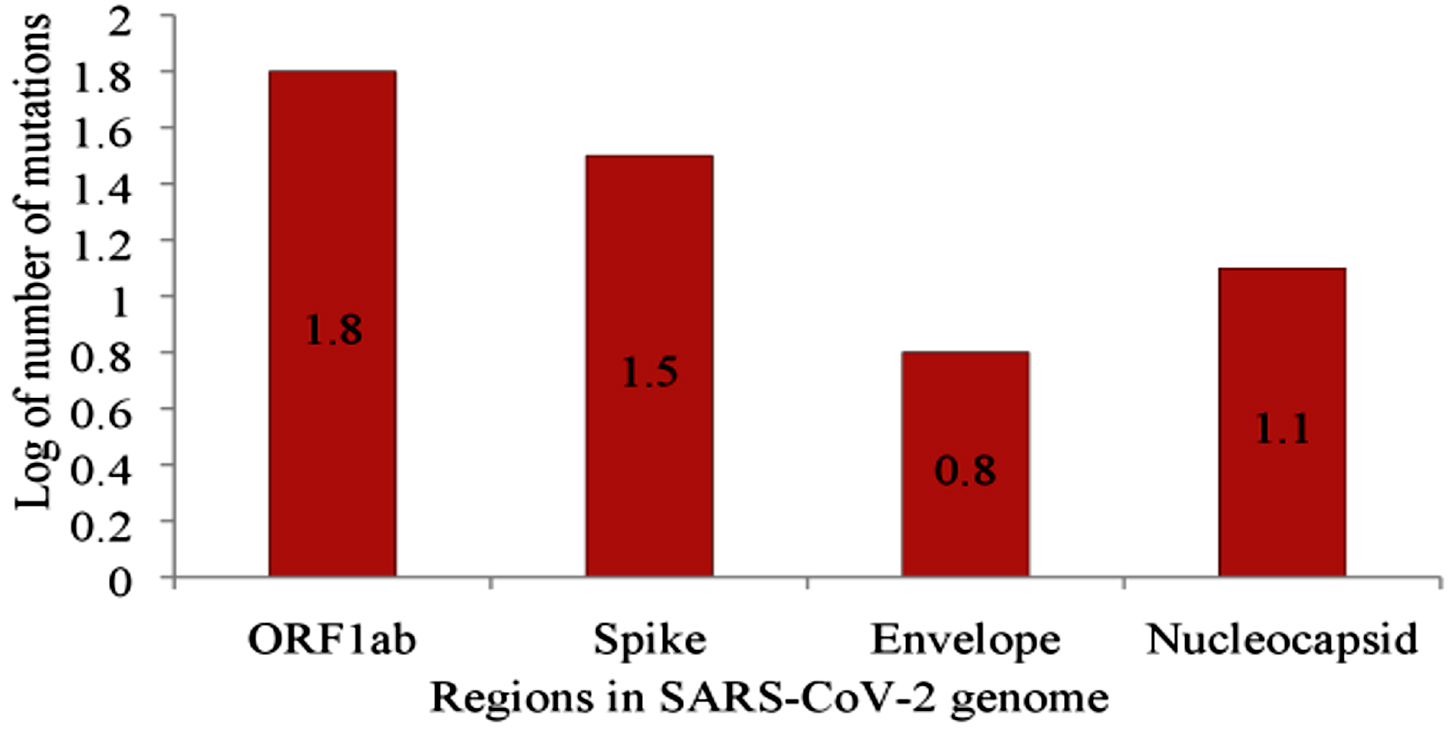
Fig. 1. Log 10 non-synonymous mutations occurring in genome of SARS-CoV-2
SARS-CoV-2 versus SARS-CoV infectivity
The S glycoprotein of CoVs is crucial for infection and stability, and determines host tropism,as well as the transmission capacity of the virus4,8. The S protein attaches to the receptor cells, allowing an entry of the virus12,35. The S gene has a subunit, S1, which has a receptor-binding domain (RBD) that binds to angiotensin-converting enzyme 2 (ACE2), allowing the S2 domain to fuse and initiatean infection. The RBDs of SARS-CoV-2 and SARS-CoV have 89.2% sequence similarity and 70%–73.7% sequence identity with several amino acids variations in key residues. SARS-CoV-2, NL63, and SARS-CoV receptors use ACE2 as their main receptor10,49, whereas MERS-CoV attaches to dipeptidyl peptidase 4.
More RBD residues are involved in binding to the receptor,and they have a longer capping loop in SARS-CoV-2 than in SARS-CoV.These structural characteristics are associated with an increased affinity for binding8. The SARS-CoV-2 S protein binds to ACE2 with significantly higher affinity than that of SARS-CoV. Therefore, it is more efficient at infecting human cells and hence, rapidly spreads in the human population8,36,38,39.The binding free energy between RBD-ACE2 in SARS-CoV-2 is lower than that of SARS-CoV and explains why it is more infectious than the latter. This indicates that it has increased stability and can withstand high temperatures8,35. The SARS-CoV-2 RBD domain has higher solubility than the SARS-CoV domain, enhancing binding to the ACE2 receptor and contributing to its high infectivity8. Besides SARS-CoV-2, the rate of protein synthesis is higher than that of HCoVs due to the overall ratios of human slow codons/di-codons present, thereby increasing the transmissibility rate. Fast-replicating viruses have a high chance of survival in the environment and have successful
infection 31.
The mutations that occur in SARS-CoV-2 residues, responsible for cross-species transmission, could be more favorable than those in SARS-CoV 31,36. Peptide insertion at the S1/S2 boundary has been attributed to SARS-CoV-2 infectivity14,16. A previous study40 reported that the S protein of SARS-CoV-2 weakly binds to its receptor, unlike SARS-CoV, justifying its severity only in immuno compromised individuals. However, this does not explain the extensive spread of SARS-CoV-2 infection within a short duration. This evidence is also supported by the trends of SARS-CoV-2 transmission, with more than 10 million people being infected within a period of six months since its outbreak.
Intermediate host
COVID-19 was first reported in nine patients with pneumonia in China in December 2019. Of the nine patients, eight had visited the Huanan wet market in Wuhan City and the other patient lived close to the market before the outbreak4,41. Huanan wet markets sell different wild animal species, including marmots, pangolins, snakes, leopard cats, bamboo rats, badgers, and hedgehogs. All of these species are susceptible to CoVs. SARS-CoV-2 is suspected to have originated from bats as it shares a 87%–96% whole genome sequence identity with batCoVs, such as RaTG13, SL-CoVZC45, and SL-CoVZXC2111,20,22. Based on the analysis of complete genomic sequences, bats are reservoirs of approximately 30 CoVs. However, during the out break period, no bats were being sold as most species had hibernated. Therefore, another organism was likely to be an intermediate host4.
Several animals, including pangolins, snakes, and turtles, are suspected to be SARS-CoV-2 intermediate hosts22,42–44. Although illegally sold, pangolins are a delicacy in China, and their scales are used in Chinese traditional medicine45.At the nucleotide level, pangolin CoVs and SARS-CoV-2 share 85.5% – 99% sequence similarity22,44,45.Pangolin-CoVs are also closely related at the whole genome level to bat CoV RaTG13 (90.55% ), SARSr-CoV ZXC21(85.65%), and bat SARSr-CoV ZC45 (85.01%)22. SARS-CoV-2 has an insertion, PRAR at the S1 and S2 sites, which induces a furin cleavage motif that is absent in pangolin CoVs. Therefore, although pangolin-CoVs are genetically closely related to SARS-COV-2 and bat CoVs, pangolins may not be intermediate hosts12,16.
Ji et al.42 reported that snakes of the species Bungarus multicinctus and Naja atra are potential intermediate hosts. However, this was refuted by another study that showed that the relative synonymous codon usage distance was smaller between SARS-CoV-2, and a frog species than these snake species23. However, they did not suggest frogs as the intermediate host but further argued that the intermediate host would most likely be a mammal. Turtles were also suspected to be a possible intermediate because of the observed interaction between RBD key aa and their ACE2. In addition, they were reported to be more common in the market12. However, more research is required to determine this possibility.
Risk factors
Several sociodemographic characteristics were associated with COVID-19. SARS-CoV-2 infects all age groups with a median age of 45–56 years. Patients aged above 45 years respond poorly to treatment; hence, they have increased fatalities5,46.COVID-19 is also more prevalent in men than in women,with a mortality rate 2.4 times higher among men than among women47–49. The high incidence observed among men could be a result of higher prevalence of smoking and movement, including international travel,compared to women in many cultures.
Behavioral characteristics, such as human–animal contact, poor hygiene and sanitation, and wet markets increase the risk of zoonotic diseases50. Smoking increases the susceptibility and death rate of SARS-CoV-2 infection5,51. ACE2 expression levels in long-term smokers are up regulated51. Normally, ACE2 is upregulated to protect the host against acute lung injury. The levels of ACE2 do not vary by sex or age. Chronic smoking triggers ACE2 secretory cell expansion, which could explain the vulnerability as SARS-CoV-2 uses the ACE2 receptor51,52.
A temperature range of 13–24°C, precipitation of less than 30mM/month, and humidity of 50%–80% favors SARS-CoV-2 infection. Lower rainfall reduces relative humidity and provides favorable conditions for the spread of pathogens causing respiratory infections53. Several studies support this hypothesis,54 reporting that an increase in temperature and absolute humidity is associated with decreased mortality after observing weather conditions in relation to the death rate for one month. The RBD-ACE2 complex results in a high entropy penalty owing to the flexibility of the RBD near the binding site. This means that it is temperature-sensitive regarding infections in humans. Therefore, an increase in temperature decreases the rate of infection, and can therefore be controlled8. The increased fatality rate of SARS-CoV-2 is correlated with the levels of particulate matter (PM) pollution (PM2.5, PM10). PM affects the outcome of respiratory diseases55.
Underlying conditions that worsen COVID-19 outcomes include diabetes mellitus, hypertension, and cardiovascular diseases35,39,46.Among patients with diabetes and cardiovascular diseases, ACE2 plays a protective role.Unfortunately, SARS-CoV-2 downregulates ACE2 protein, resulting in severe clinical outcomes in these patients. Both, patients with respiratory diseases,as well as healthy individuals, are susceptible to SARS-CoV-2 infection 51. Both healthy individuals and those with chronic respiratory diseases showed similar ACE2 expression levels51. The recurrence of positive SARS-CoV-2 RNA predictors includes high levels of IL-6, elevated lymphocyte counts, and lung consolidation features upon hospital admission11,39,51. The transmission of SARS-CoV-2 in countries with malaria incidence is limited 56. This could be a result of the wide use of antimalarial drugs, especially chloroquine. Chloroquine is a broad-spectrum antiviral agent that increases endosomal pH, which is essential for enhancing the fusion of the virus and host cells. It inhibits the uncoating and glycosylation of many viruses.Chloroquine in combination with other antiviral agents was reported to be effective in controlling COVID-1956,57.
The outbreaks
COVID-19 was first reported on December 31, 2019, SARS on November 1, 2002, and MERS on September 20, 2012 but was traced back to April 2012. Therefore, this study focused on the first six months of the outbreak: COVID-19 on June30,2020, SARS upto April 30, 2003, and MERS upto February 20, 2013. By June 30, 2020, approximately 10 million people had contracted COVID-19 resulting in approximately 500,000 fatalities. This is more than 1,000 times as many people that contracted SARS and MERS and died during the six months after their outbreaks (Table 3). Of the three outbreaks, COVID-19 is the most extensively spread and was reported in over 200 countries within the first six months of its emergence; in contrast, MERS and SARS were reported in 5 and 28 countries, respectively.However, the COVID-19 mortality rate within the first six months of the outbreak was 4.9%, indicating that it is less aggressive than SARS (6.6%) and MERS (65%). The etiological agents for these outbreaks have been linked to bats with different intermediate hosts.
Table (3):
The first six months of COV1D-19, MERS and SARS disease outbreaks.
Organism |
SARS-COV-2 |
SARS-COV |
MERS-CoV |
References |
|---|---|---|---|---|
Disease |
Covid-19 |
SARS |
MERS |
2,3 |
First case presented in |
China |
China |
Saudi Arabia |
2,3 |
Overall Case fatality in % |
4.9 |
6.6 |
65 |
|
Incubation Period in days |
2-14 |
2-11 |
2-13 |
3,46 |
Median to time of death in days |
14 |
23 |
14 |
3,46 |
Cases |
10 185 374 |
5663 |
17 |
68-70 |
Deaths |
503862 |
372 |
11 |
68-70 |
Transmission mode |
Droplets, close contact |
Droplets, close contact |
Droplets, close contact |
3,39 |
Human-human transmission |
Efficient |
Efficient |
Limited |
71 |
Pandemic potential |
Yes |
Yes |
No |
71 |
Distribution in countries |
213 |
28 |
5 |
68-70 |
Reservoir |
Bats |
Rhinolophussinicus |
Bats and alpacas |
3,39 |
Intermediate host |
Pangolins (to be confirmed) |
Palm civets |
Dromedary camel |
3,39,68-70 |
We reviewed studies that reported the genomes, mutations and epidemiology of the pathogen responsible for COVID-19 and the number of selected CoVs. The SARS-CoV-2 genomeis highly similar to the strain RaTG13 of bat origin. This was followed by pangolins based on amino acid similarities of the whole genome, S, M, E, and N. Mutations occurred at a high frequency in important proteins, namely the S, N, and replicase. ORFs 6, 7, and 10 were highly conserved. Insights into the sequence variations, especially important genes, such as the S glycoprotein, are important for understanding the biology of SARS-CoV-2 infection. This knowledge could be useful for antiviral treatment and vaccine development. Although SARS-CoV-2 and SARS-CoV use the same receptor in humans, the former has high binding affinity to the receptor. This results in high infectivity and transmissibility rates. Despite evidence of a close relationship between pangolins and SARS-CoV-2 in most studies, there remains an uncertainty regarding their identification as intermediate hosts. The risk factors for COVID-19 have been defined and are similar to those of other respiratory diseases; smoking and air pollution have been associated with diseasese verity. However, it has not been associated with cold weather, and this could partly explain the mortality variations in different regions. SARS-CoV-2 is highly transmissible and infectious, with a low mortality rate. We recommend further research to identify SARS-CoV-2 intermediate hosts to avoid future spillover, and to establish the significance of different mutations.
ACKNOWLEDGMENTS
None.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest
AUTHORS’ CONTRIBUTION
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
FUNDING
None.
ETHICS STATEMENT
Not applicable.
AVAILABILITY OF DATA
All data generated or analyzed during this study are included in the manuscript.
- Gorbalenya AE, Baker SC, Baric RS, et al. The species Severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol. 2020;5(4):536-544.
Crossref - World Health Organization (WHO). Novel coronavirus – China 11-fev 2020. WHO Bull. 2020;(February). https://www.who.int/docs/default-source/coronaviruse/situation-reports/20200211-sitrep-22-ncov.pdf?sfvrsn=fb6d49b1_2.
- Su S, Wong G, Shi W, et al. Epidemiology , Genetic Recombination , and Pathogenesis of Coronaviruses. Trends Microbiol. 2016;24(6):490-502.
Crossref - Lu R, Zhao X, Li J, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395(10224):565-574.
Crossref - Guo YR, Cao QD, Hong ZS, et al. The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak- A n update on the status. Mil Med Res. 2020;7(1):1-10.
Crossref - Kim J-S, Jang J-H, Kim J-M, Chung Y-S, Yoo C-K, Han M-G. Genome-Wide Identification and Characterization of Point Mutations in the SARS-CoV-2 Genome. Osong Public Heal Res Perspect. 2020;11(3):101-111.
Crossref - Cui H, Gao Z, Liu M, et al. Structural genomics and interactomics of 2019 Wuhan novel coronavirus, 2019-nCoV, indicate evolutionary conserved functional regions of viral proteins. bioRxiv. 2020:2020.02.10.942136.
Crossref - He J, Tao H, Yan Y, Huang SY, Xiao Y. Molecular mechanism of evolution and human infection with SARS-CoV-2. Viruses. 2020;12(4):1-19.
Crossref - Ivair José Morais Júnior, Richard Costa Polveiro, Gabriel Medeiros Souza, Daniel Inserra Bortolin, Flávio Tetsuo Sassaki ATML, Morais IJ, et al. The global population of SARS-CoV-2 is composed of six major subtypes. Sci Rep. 2020;10(1):2-10.
Crossref - Gao Y, Yan L, Huang Y, et al. Structure of RNA-dependent RNA polymerase from 2019-nCoV, a major antiviral drug target. bioRxiv. 2020:2020.03.16.993386.
Crossref - Chen Y, Liu Q, Guo D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J Med Virol. 2020;92(4):418-423.
Crossref - Liu Z, Xiao X, Wei X, et al. Composition and divergence of coronavirus spike proteins and host ACE2 receptors predict potential intermediate hosts of SARS-CoV-2. J Med Virol. 2020;(February):1-7.
Crossref - Li X, Wang W, Zhao X, et al. Transmission dynamics and evolutionary history of 2019-nCoV. J Med Virol. 2020;92(5):501-511.
Crossref - Walls Alexandra C., Park Young-Jun, Tortorici M. Alejandra, Wall Abigail, McGuire Andrew T. VD. Structure , Function , and Antigenicity of the SARS-. Cell. 2020;180:1-12.
- Tiwari M, Mishra D. Investigating the genomic landscape of novel coronavirus (2019-nCoV) to identify non- synonymous mutations for use in diagnosis and drug design Manish. 2020:1-5.
Crossref - Wu A, Peng Y, Huang B, et al. Genome Composition and Divergence of the Novel Coronavirus (2019-nCoV) Originating in China. Cell Host Microbe. 2020;27(3):325-328.
Crossref - MacKenzie JS, Smith DW. COVID-19: A novel zoonotic disease caused by a coronavirus from China: What we know and what we don’t. Microbiol Aust. 2020;41(1):45-50.
Crossref - Tang X, Wu C, Li X, et al. On the origin and continuing evolution of SARS-CoV-2. Natl Sci Rev. 2020;7(6):1012-1023.
Crossref - Malik YS, Sircar S, Bhat S, et al. Emerging novel coronavirus (2019-nCoV)—current scenario, evolutionary perspective based on genome analysis and recent developments. Vet Q. 2020;40(1):68-76.
Crossref - Fahmi M, Kubota Y, Ito M. Nonstructural proteins NS7b and NS8 are likely to be phylogenetically associated with evolution of 2019-nCoV. Infect Genet Evol. 2020;81(March):104272.
Crossref - Benvenuto D, Demir AB, Giovanetti M, et al. Evidence for mutations in SARS‐CoV‐2 Italian isolates potentially affecting virus transmission. J Med Virol. 2020:0-3.
Crossref - Zhang T, Wu Q, Zhang Z. Pangolin homology associated with 2019-nCoV. bioRxiv. 2020:2020.02.19.950253.
Crossref - Zhang C, Zheng W, Huang X, Bell EW, Zhou X, Zhang Y. Protein Structure and Sequence Reanalysis of 2019-nCoV Genome Refutes Snakes as Its Intermediate Host and the Unique Similarity between Its Spike Protein Insertions and HIV-1. J Proteome Res. 2020;19(4):1351-1360.
Crossref - Wan Y, Shang J, Graham R, Baric RS, Li F. Receptor recognition by novel coronavirus from Wuhan: An analysis based on decade-long structural studies of SARS. J Virol. 2020;94(e00127-20):1-9.
Crossref - Dilucca M, Pavlopoulou A. Analysis of codon usage and evolutionary rates of the 2019-nCoV genes. bioRxiv. 2020;7:2020.03.25.006569.
Crossref - Pachetti M, Marini B, Benedetti F, et al. Emerging SARS-CoV-2 mutation hot spots include a novel RNA-dependent-RNA polymerase variant. J Transl Med. 2020;18(1):1-9.
Crossref - van Dorp L, Acman M, Richard D, et al. Emergence of genomic diversity and recurrent mutations in SARS-CoV-2. Infect Genet Evol. 2020;83(May):104351.
Crossref - Lokman SM, Rasheduzzaman M, Salauddin A, et al. Exploring the genomic and proteomic variations of SARS-CoV-2 spike glycoprotein: A computational biology approach. Infect Genet Evol. 2020;84.
Crossref - Yin C. Genotyping coronavirus SARS-CoV-2: methods and implications. Genomics. 2020;(January).
Crossref - Ceraolo C, Giorgi FM. Genomic variance of the 2019-nCoV coronavirus. J Med Virol. 2020;92(5):522-528.
Crossref - Yang CW, Chen MF. Composition of human-specific slow codons and slow di-codons in SARS-CoV and 2019-nCoV are lower than other coronaviruses suggesting a faster protein synthesis rate of SARS-CoV and 2019-nCoV. J Microbiol Immunol Infect. 2020;(xxxx):0-5.
Crossref - GitHub – nextstrain/ncov: Nextstrain build for novel coronavirus SARS-CoV-2. https://github.com/nextstrain/ncov.
- Ugurel OM, Ata, Oguz, Turgut-Balik D. An updated analysis of variations in SARS-CoV-2 genome. Turkish J Biol. 2020;44:157-167.
Crossref - Phan T. Genetic diversity and evolution of SARS-CoV-2. Infect Genet Evol. 2020;81(January).
Crossref - He F, Deng Y, Li W. Coronavirus disease 2019: What we know? J Med Virol. 2020;2019:0-2.
Crossref - Ortega JT, Serrano ML, Pujol FH, Rangel HR. Role of changes in SARS-COV-2 spike protein in the interaction with the human ACE2 Recepter: An in silico analysis. EXCLI J. 2020;19(19):410-417.
Crossref - Cao Y, Li L, Feng Z, et al. Comparative genetic analysis of the novel coronavirus (2019-nCoV/SARS-CoV-2) receptor ACE2 in different populations. Cell Discov. 2020;6(1):4-7.
Crossref - Lan J, Ge J, Yu J, et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020.
Crossref - Xu J, Zhao S, Teng T, et al. Systematic comparison of two animal-to-human transmitted human coronaviruses: SARS-CoV-2 and SARS-CoV. Viruses. 2020;12(2).
Crossref - Dong N, Yang X, Ye L, Chen K, Chan EW-C, Chen S. Genomic and protein structure modelling analysis depicts the origin and pathogenicity of 2019-nCoV, a new coronavirus which caused a pneumonia outbreak in Wuhan, China. F1000Research. 2020;9:121.
Crossref - Zhou P, Yang X, Wang X, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(March).
Crossref - Ji W, Wang W, Zhao X, Zai J, Li X. Homologous recombination within the spike glycoprotein of the newly identified coronavirus may boost cross-species transmission from snake to human. J Med Virol. 2020;92(4):20200122.
Crossref - Li X, Zai J, Zhao Q, et al. Evolutionary history, potential intermediate animal host, and cross-species analyses of SARS-CoV-2. J Med Virol. 2020:1-10.
Crossref - Liu Ping, Jiang Jing-Zhe, Wan Xiu-Feng, Hua Yan, Wang Xiaohu, Hou Fanghui, Chen Jing, Zou Jiejian CJ. Are pangolins the intermediate host of the 2019 novel coronavirus (2019-nCoV) ? bioRxiv Prepr. 2020:1-9.
Crossref - Lam TT-Y, Shum MH-H, Zhu H-C, et al. Identifying SARS-CoV-2 related coronaviruses in Malayan pangolins. Nature. 2020:1-6.
Crossref - Rothan HA, Byrareddy SN. The epidemiology and pathogenesis of coronavirus disease (COVID-19) outbreak. J Autoimmun. 2020;(February):18-21.
Crossref - Coronavirus Age, Sex, Demographics (COVID-19) – WorldometerNo Title. https://www.worldometers.info/coronavirus/coronavirus-age-sex-demographics/.
- Jin JM, Bai P, He W, et al. Gender Differences in Patients With COVID-19: Focus on Severity and Mortality. Front Public Heal. 2020;8(April):1-6.
Crossref - World Health Organization (WHO). Gender and COVID-19. 2020;(May):6-9.
- Li HY, Zhu GJ, Zhang YZ, et al. A qualitative study of zoonotic risk factors among rural communities in southern China. Int Health. 2019;12(2):77-85.
Crossref - He X, Zhang L, Ran Q, et al. Integrative Bioinformatics Analysis Provides Insight into the Molecular Mechanisms of 2019-nCoV. 2020.
Crossref - Smith JC, Sausville EL, Girish V, et al. Cigarette Smoke Exposure and Inflammatory Signaling Increase the Expression of the SARS-CoV- 2 Receptor ACE2 in the Respiratory Tract. Dev Cell. 2020;53(5):514-529.e3.
Crossref - Bu J, Peng D-D, Xiao H, et al. Analysis of meteorological conditions and prediction of epidemic trend of 2019-nCoV infection in 2020. medRxiv. 2020;(May):2020.02.13.20022715.
Crossref - Ma Y, Zhao Y, Liu J, et al. Effects of temperature variation and humidity on the death of COVID-19 in Wuhan, China. Sci Total Environ. 2020;724:138226.
Crossref - Yao Y, Pan J, Liu Z, et al. Temporal Association Between Particulate Matter Pollution and Case Fatality Rate of COVID-19 in Wuhan, China. medRxiv. 2020:2020.04.09.20049924.
Crossref - Bajaj A, Purohit HJ. Understanding SARS-CoV-2: Genetic Diversity, Transmission and Cure in Human. Indian J Microbiol. 2020.
Crossref - Lai CC, Shih TP, Ko WC, Tang HJ, Hsueh PR. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease-2019 (COVID-19): The epidemic and the challenges. Int J Antimicrob Agents. 2020;55(3):105924.
Crossref - Liu Ping, Jiang Jing-Zhe, Wan Xiu-Feng, Hua Yan, Wang Xiaohu, Hou Fanghui, Chen Jing, Zou Jiejian CJ, Liu P, Jiang JZ, et al. Are pangolins the intermediate host of the 2019 novel coronavirus (SARS-CoV-2)? PLoS Pathog. 2020;16(5):1-13.
Crossref - Xiao K, Zhai J, Feng Y, et al. Isolation and Characterization of 2019-nCoV-like Coronavirus from Malayan Pangolins. bioRxiv. 2020:2020.02.17.951335.
Crossref - Xiao K, Zhai J, Feng Y, et al. Isolation of SARS-CoV-2-related coronavirus from Malayan pangolins. Nature. 2020;583(7815):286-289.
Crossref - Xu Y. Genetic diversity and potential recombination between ferret coronaviruses from European and American lineages. J Infect. 2020;80(3):350-371.
Crossref - Zhou Y, Zhang S, Chen J, Wan C, Zhao W, Zhang B. Analysis of variation and evolution of SARS-CoV-2 genome. J South Med Univ. 2020;40(2):152-158.
Crossref - Poterico JA, Mestanza O. Genetic variants and source of introduction of SARS‐CoV‐2 in South America. J Med Virol. 2020:0-1.
Crossref - Chaw SM, Tai JH, Chen SL, et al. The origin and underlying driving forces of the SARS-CoV-2 outbreak. J Biomed Sci. 2020;27(1):73.
Crossref - Maitra A, Sarkar MC, Raheja H, et al. Mutations in SARS-CoV-2 viral RNA identified in Eastern India: Possible implications for the ongoing outbreak in India and impact on viral structure and host susceptibility. J Biosci. 2020;45(1):1-18.
Crossref - Wang C, Liu Z, Chen Z, et al. The establishment of reference sequence for SARS-CoV-2 and variation analysis. J Med Virol. 2020;92(6):667-674.
Crossref - Benvenuto D, Angeletti S, Giovanetti M, et al. Evolutionary analysis of SARS-CoV-2: how mutation of Non-Structural Protein 6 (NSP6) could affect viral autophagy. J Infect. 2020;81:e24-e27.
Crossref - WHO | Update 43 – Situation in China: more cases than the rest of the world combinedNo Title. https://www.who.int/csr/sars/archive/2003_04_30/en/.
- World Health Organization (WHO). Coronavirus disease, Situation Report -162. 2020;(June).
- Zumla AI, Memish ZA. Middle East respiratory syndrome coronavirus: epidemic potential or a storm in a teacup? Alimuddin. Eur Respir J. 2014;43:1243-1248.
Crossref - Lee Ping-Ing, Hsueh P-R. Emerging threats from zoonotic coronaviruses-from SARS and MERS to 2019-nCoV. J Microbiol Immunol Infect. 2020;(January):19-21.
© The Author(s) 2021. Open Access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License which permits unrestricted use, sharing, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.