


ISSN: 0973-7510
E-ISSN: 2581-690X
A bacterium isolated from Lonar Soda lake sample was tested for its cellulolytic activity, which was showing more activity on cellobiose as substrate than CMC and sugarcane bagasse. CMCase activity was highest at 72 h of incubation and declined on further increase in the time of incubation. Experiments were conducted to determine the effect of different carbon sources on CMCase production. Overall, the bacterium showed the highest CMCase activity; i.e., 0.45 IU mL-1 at 72 h, when cellobiose was used as a carbon source. Genomic sequencing of this strain B14 was performed which was showing an assembly length of 5.39 Mb. Gene ontology analysis assignied 3907, 3012 and 1003 genes associated with known molecular functions, biological processes and cellular components, respectively. The total protein coding genes present were 5477, amounting to 97.89% of the genome size. Of these, 4150 genes were protein coding genes with known functions and 1327 genes were protein coding genes with some unknown functions. The gene annotation using PANTHER identified 9 different sequences across all the contigs which belonged to Glycosyl hydrolase family13. Of these, only one gene was identified as a a-amylase, the remaining 8 genes are potentially novel genes belonging to the family present in the strain B14.
Cellulolytic bacterium, Bacillus aryabhattai B14, Lonar soda lake, Comparative genomic analysis.
Interest in bioenergy has been sharply increasing in recent years due to the necessity of sustainable economies and clean environments (Lynd et al., 2008). Cellulose and hemicellulose are the most abundant biomasses on earth, and therefore have the greatest potential to resolve both the energetic and environmental demands of bioenergy (Sánchez and Cardona 2008). Cellulolytic organisms are ubiquitous in nature. Both fungi and bacteria have been heavily exploited for their abilities to produce a wide variety of cellulases and hemicellulases. Bacteria have some advantages over fungi in certain aspects. In particular, they usually have a higher growth rate allowing for faster production of recombinant enzymes (Maki et al., 2009). In addition, some glycoside hydrolases from bacteria are assembled in multi-enzyme complexes that provide increased synergy, stability and catalytic efficiency (Hou et al., 2006; Jiang et al., 2006; Waeonukul et al., 2009), while others display modular architecture (Cann et al., 1999; Zhang et al., 2014) or are multifunctional, harboring both endoglucanase and xylanase activities in the same polypeptide (Pérez-Avalos et al., 2008). Finally, cellulolytic bacteria have been isolated from harsh climate conditions (Soares et al., 2012). Hence, their enzymes are more stable under extreme conditions (high temperature, extremes of pH) that may occur during bioconvertion processes and this may increase the overall efficiency of the enzymatic hydrolysis and fermentation (Maki et al., 2009).
Over the years, culturable cellulolytic bacteria have been isolated from a wide variety of environments, such as, compost piles, decaying plant material originating from agricultural wastes, faeces of ruminants, soil, gastrointestinal tract of insects and from extreme environments, such as, hot springs (Doi, 2008). Screening for cellulase producing organisms may be accomplished through medium enrichment with crystalline cellulose, followed by 16S rRNA sequencing to determine the composition of the bacterial communities present and evaluate whether families containing cellulolytic species are present. Strains with cellulolytic potential can be isolated by subsequent subcultures in the enriched culture medium containing cellulose as carbon source (Maki et al., 2009; Rastogi et al., 2009). Alternatively, screening of cellulases producing by bacterial isolates may be accomplished by their cultivation in solid media containing carboxymethylcellulose (CMC) as sole carbon source, followed by Congo Red staining (Hankin and Anagnostakis, 1977). CMC is a highly specific substrate for endo-acting cellulases, as its structure has been engineered to decrystallize cellulose and create amorphous sites that are ideal for endoglucanase action, called CMCase, that cleaves intramolecular â-1,4-glucosidic bonds randomly, resulting in a dramatic reduction of the degree of polymerization and specific viscosity of CMC (Zhang et al., 2006). Although CMC has become a commonly used surrogate for cellulose, as many had associated whole cellulase activity with CMC hydrolysis (Liang et al., 2014), cellobiohydrolases are shown to be dominant in the degradation of crystalline (e.g., Avicel) and not soluble (e.g., CMC) cellulose (Zhang et al., 2006).
Isolation and identification
The samples collected from the vicinity of the Lonar soda lake were inoculated in 50 mL of basal salt media gL-1(NaNO3, 2.5; MgSO4, 0.2; NaCl, 0.2; CaCl2 .6H2O, 0.1) containing filter paper (Whatman filter paper no.1, each of 5 mg and area 0.49 cm2) for isolating the cellulolytic microorganisms. A control was prepared with basal salt media and Whatman filter paper but without the sample for checking the degradation/disappearance of filter paper. The tubes were incubated aerobically on a shaking incubator at 180 rpm and 37 °C. After the filter paper was visibly degraded, the sample was serial diluted and 10 µL of the dilution was transferred on to solid medium (CMC agar ). The CMC agar medium contained g/L-1: NaNO3, 2.5; MgSO4, 0.2; NaCl, 0.2; CaCl2 .6H2O, 0.1; agar, 20; CMC 1. All the plates were incubated at 37°C until colonies were visible. The identification of isolates for their cellulolytic potential was performed using the Congo-Red overlay method (Teather and Wood 1982). For this, plates were flooded with 0.1% Congo red (Sigma-Aldrich) for 10-15 min before de-staining with 1M NaCl solution for 15-20 min for several times or until the clear zones around the colonies were visualized. Colonies showing discoloration of Congo red were taken as positive cellulose-degrading microbial colonies. The microbes, which produced clear zones, were identified based on the cultural, morphological and biochemical characteristics and using Bergey’s Manual of Systemic Bacteriology (Holt et al., 1994).
Production of Cellulase
The sugarcane bagasse samples were dried at 60°C for 24 h. Then they were ground to a powder in a blender. The ground bagasse was sieved and it was autoclaved twice (121°C for 15 min). The medium used for production of the cellulase contained the following components (g/L): sugarcane bagasse powder 10; NaNO3 0.5; K2HPO4 1.0; MgSO4.7H2O 0.5; FeSO4.7H2O 0.01 and yeast extract 1.0 at pH 7.0. The inoculum medium was the same medium with 1% glucose added. Cellulase production was carried out in 250 mL conical flasks containing 100 mL of the described medium. These were then inoculated with 1% (v/v) of inoculum culture and incubated for 18 h at 37°C under shaking at 150 rpm. After 72 h of incubation, the contents of the fermented flasks were subjected to centrifugation at 9,000 g for 15 min at 4 °C and the clear cell-free supernatant (crude extract) was collected and stored at 4 °C before enzymatic assay. Using the same method, other carbon sources (glucose, cellobiose and CMC) were evaluated.
Enzymatic Assay
CMCase activity was determined by estimating the reducing sugars produced in 10 min from a mixture of enzyme solution (0.25 mL) and 0.25 mL of a 4.5% CMC (Sigma) solution made in 100 mM sodium phosphate buffer at pH 7.0 and 50 °C. After incubation, the concentration of reducing sugars was determined using the dinitrosalicylic acid (DNS) method. One unit of CMCase was defined as the amount of enzyme that catalyzes and releases 1ìmol of reducing sugar equivalent per minute under the specified assay conditions. All activity measurements were repeated three times.
DNA isolation and sequencing
DNA isolation from pure cultures was performed using the Easy-DNA kit (Invitrogen) with an additional pretreatment step. Initially, the cells were inoculated onto a blood agar plate and were incubated overnight at 37°C. A single colony was then inoculated into 10 ml brain heart infusion (BHI) broth and incubated overnight at 37°C with gentle shaking (75 rpm). The 10-ml overnight culture was centrifuged at 5,000 × g for 10 min and resuspended in 200 ìl phosphate-buffered saline. Lysozyme (30 µl of a 10 g/liter suspension, to a final concentration of 1.3 g/liter) was added to this mixture and the cells were incubated for 20 min at 37°C. After incubation, 30 µl of 10% sodium dodecyl sulfate was added and the tubes were gently mixed. Finally, 15 µl of proteinase K (20 g/liter) was added and the samples were incubated for 20 min at 37°C. DNA was then purified as described in the Easy-DNA protocol. The quantification of the genomic DNA sample was done using QUBIT fluorometer. The library was prepared using NEBNext® Ultra™ DNA II Library Prep Kit for Illumina® with Illumina standardized protocol (Catalog No: NEB E7370). The final enriched libraries were further validated for quality on Agilent Bioanalyzer using DNA High Sensitivity chip and for quantification on real time PCR (KAPA Library Quantification kit). The PE libraries were sequenced on Illumina NextSeq 500 platform, using TruSeq PE Kit (2 x 150bp) with 50X coverage per sample.
Data analysis
The quality of raw reads of Illumina sequencing was checked for the ambiguous bases, Phred score >Q30, read length, nucleotide base content and other parameters by using FASTQC. The quality processed Illumina paired end reads were used for De novo assembly with three different assembly tools: CLC bio Genomics Work Bench (version: 9.5.2). Gene prediction was done using Glimmer version 3.02. Gene annotation was carried out with Blast2GO and BASys along with propriety tools. A number of databases were scanned to retrieve the gene annotation, such as, BlastProDom, FPrintScan, PANTHER, Pfam, Smart and SuperFamily.
Isolation, identification and selection of cellulolytic bacterial strains
The enrichment and selective isolation of isolates on CMC agar lead to identification of 6 strains of bacteria. They include a few Bacillus spp. and Stenotrophomonas spp. Among these, the isolates showing prominent zone formation on the CMC agar after congo-red staining was a Bacillus sp. B14. Based on 16S rDNA sequencing, biochemical and physiological characteristics it was identified as a strain of previously described Bacillus aryabhattai (Table 1).
Table (1):
Biochemical characterization of strain B14.
Characteristic |
Strain B14 |
|---|---|
Colony pigmentation |
Creamy |
Motility |
+ |
Utilization of: |
|
Lactose |
+ |
Xylose |
+ |
Maltose |
+ |
Fructose |
+ |
Dextrose |
+ |
Galactose |
+ |
Raffinose |
+ |
Trihalose |
+ |
Melobiose |
+ |
Sucrose |
+ |
L-arabinose |
+ |
Mannose |
– |
Inulin |
+ |
Sodium gluconate |
+ |
Glycerol |
+ |
Salicin |
– |
Dulcitol |
– |
Inositol |
– |
Sorbitol |
– |
Mannitol |
+ |
Adonitol |
– |
Arabitol |
– |
Erythritol |
– |
α-methyl-D-glucoside |
– |
Rhamnose |
– |
Cellobiose |
+ |
Melezitose |
+ |
α-methyl-D-mannoside |
– |
Xylitol |
– |
D-arabinose |
+ |
Citrate |
– |
Malonate |
– |
Sorbose |
– |
ONPG hydrolysis |
+ |
Esculin hydrolysis |
+ |
Cellulase production and assay
Several cellulases have been found in different members of the genus Bacillus (Rawat and Tiwari, 2012). Bacillus aryabhattai B14 showed clear zones on CMC agar plates followed by staining with 1% Congo red solution, indicating that it secretes CMCase. The CMCase production by Bacillus aryabhattai B14 was high when the cell population entered into the stationary phase, suggesting that enzyme secretion is not growth-associated. CMCase activity was highest at 72 h of incubation and declined on further increase in the time of incubation. Experiments were conducted to determine the effect of different carbon sources on CMCase production. Overall, cellobiose showed the highest CMCase activity; i.e., 0.45 IU mL-1 at 72 h (Fig. 1).
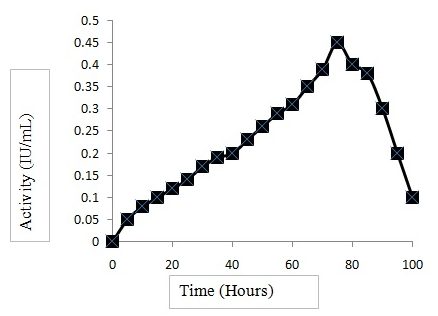 Fig.1. CMCase activity of B. aryabhattai B14
Fig.1. CMCase activity of B. aryabhattai B14Genome sequencing and data analysis
Though the strain B14 was closely related to a few previously isolated Bacillus strains, it was isolated from a different environment i.e. a slightly halophilc alkaline lake, hence it was considered for whole genome sequencing which was performed at Bangalore genomics centre, Bangalore, India. The metrics, such as, N50, longest contigs and sum of contig length (total bases) indicate that the present De novo assembly has a good quality (Table 2). Based on the assembled short reads by different assemblers, the genome size of Bacillus sp. is 5.39 Mb. The Table 3 summarizes the assembled individual nucleotide (A, T, G, C and N) count. The count of the A and T are more when compared to G and C. From the assembly statistics, we can infer that Bacillus sp. genome is AT rich.
Table (2):
De novo Assembly and different metrics.
Attributes |
CLC Workbench |
|---|---|
N50 |
563,631 bp |
Longest Contig |
1,740,531 bp |
Total Contigs |
146 |
Assembly Length |
5,396,710 bp |
Gap Length |
918 bp |
Table (3):
De novo Assembly Nucleotide distribution.
Nucleotide |
CLC Workbench |
|---|---|
Adenine(A) |
1,677,994 bp |
Guanine(G) |
1,023,405 bp |
Cytosine(C) |
1,020,064 bp |
Thymine(T) |
1,674,329 bp |
Gene ontology analysis
Gene ontology analysis assigned 3907, 3012 and 1003 genes associated with known molecular functions, biological processes and cellular components respectively as illustrated by the Figure 2.
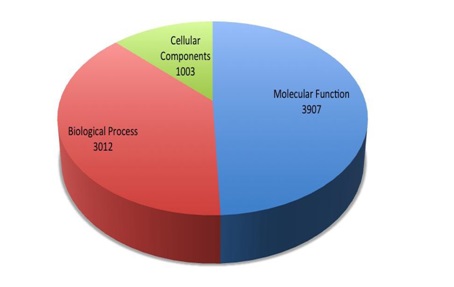
Fig. 2. Genes associated with the Molecular Functions, Biological Process and Cellular Components
The sequence distributions for each of these three GO terms are illustrated (Figs. 3-5). Totally 1003 genes were associated for cellular component, out of which 393 genes are associated with integral component of the membrane, 198 with cytoplasm and 111 are found in macromolecular complexes.
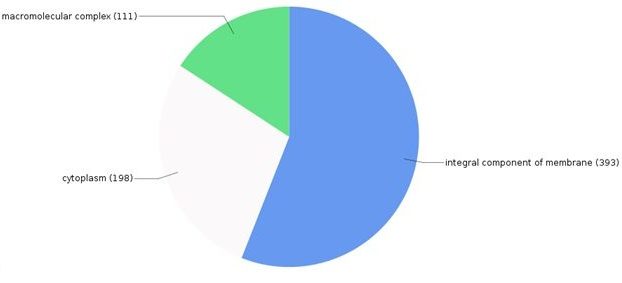 Fig. 3. Breakdown of proteins associated with various cellular components
Fig. 3. Breakdown of proteins associated with various cellular components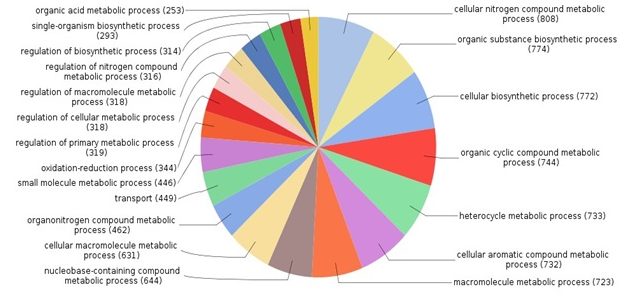
Fig. 4. Breakdown of distributions of proteins according to various biological processes
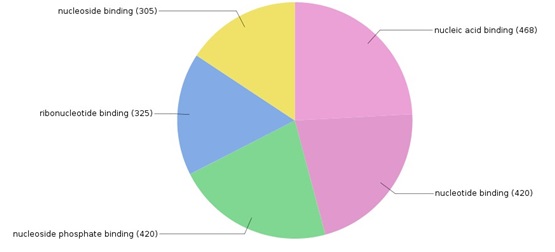 Fig. 5. Distribution of genes associated to various molecular functions
Fig. 5. Distribution of genes associated to various molecular functions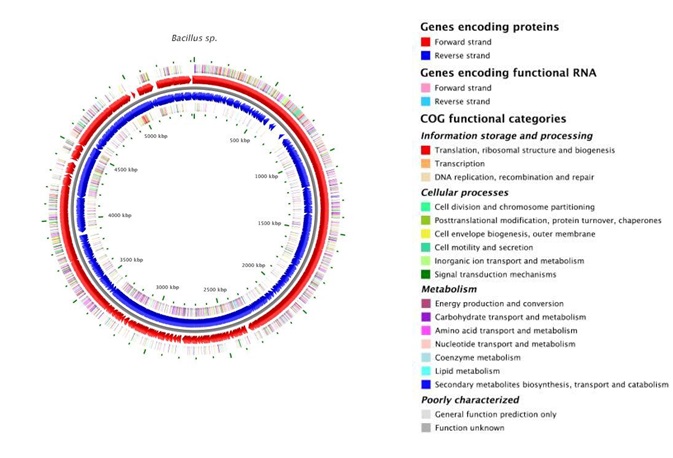
Fig. 6. Annotation map showing Genes encoding proteins
Gene annotation by BASys
A subsequent annotation was performed with BASys version 1.0 (Bacterial Annotation System). BASys is custom designed for in-depth annotation exclusively for bacterial genomic sequences. Taking our species into accordance BASys was chosen to validate the annotation previously performed by Blast2GO pipeline. The results of BASys are almost same as Blast2GO.
Genome features
The genome of B. aryabhattai strain B14 was uploaded to Integrated Microbial Genomics and Microbiome Samples website of JGI institute, USA (https://img.jgi.doe.gov/cgi-bin/mer/main.cgi) with the genome ID 2713897452. The genome identified the strain B14 as an auxotroph for L-lysine, L-phenylalanine, L-tyrosine, L-trypthopan, L-phenylalanine, L-isoleucine, L-leucine, L-serine and L-valine. The strain showed protoptrophic growth for L- aspartate, L-alanine, L-glutamate, glycine, L-asparagine and L-glutamine. The genome G + C mol % was 37.84% is agreement with most of the Bacillus sp. The total protein coding genes present were 5477, amounting to 97.89% of the genome size. Of these, 4150 genes were protein coding genes with known functions and 1327 genes were protein coding genes with some unknown function, thus making it a versatile strain of the species Bacillus aryabhattai with more number of novel genes when compared with genomes of all the sequenced strains from the species using CMGbiotools (data not shown).
The gene annotation using PANTHER identified 9 different sequences across all the contigs which belonged to glycosyl hydrolase family13. Of these, only one gene was identified as a a-amylase, the remaining 8 genes are potentially novel genes belonging to the family present in the strain B14. The identification and characterization of these genes is a prospective future work.
The draft sequence of Bacillus aryabhattai strain B14 obtained in this Whole Genome Shotgun project has been deposited at GenBank under the accession no. MVJH00000000. The version described in this paper is the first version, with accession, no. MVJH00000000. The Genomes on Line Database (GOLD) ID is PRJNA374936.
ACKNOWLEDGMENTS
MT and SVR thank Department of Science & Technology, Government of India, for providing FTSYS project grants (SB/FT/LS-320/2012 and SB/FT/LS-115/2012, respectively).
- Cann IK, Kocherginskaya S, King M.R, White BA, Mackie RI, Molecular cloning, sequencing, and expression of a novel multidomain mannanase gene from Thermoanaerobacterium polysaccharolyticum. J. Bacteriol., 1999; 181: 1643–1651.
- Doi RH, Cellulases of mesophilic microorganisms: cellulosome and noncellulosome producers. Ann. N.Y. Acad. Sci., 2008; 1125: 267–279.
- Hankin L, Anagnostakis SL, Solid media containing carboxymethylcellulose to detect CX cellulose activity of micro-organisms. J. Gen. Microbiol., 1977; 98: 109–115.
- Holt JG, Krieg NR, Sneath PHA, Staley JT, Williams ST. Bergey’s Manual of Determinative Bacteriology. 9th ed. Baltimore: Williams & Wilkins; 1994; p.786.
- Hou P, Li Y, Wu B, Yan Z, Yan B, Gao P, Cellulolytic complex exists in cellulolytic Myxobacterium Sorangium. Enzyme Microb. Technol., 2006; 38: 273–278.
- Jiang Z, Dang W, Yan Q, Zhai Q, Li L, Kusakabe I, Subunit composition of a large xylanolytic complex (xylanosome) from Streptomyces olivaceoviridis E-86. J. Biotechnol., 2006; 126: 304–312.
- Liang YL, Zhang Z, Wu M, Wu Y, Feng JX, Isolation, screening, and identification of cellulolytic bacteria from natural reserves in the subtropical region of China and optimization of cellulase production by Paenibacillus terrae ME27-1. Biomed. Res. Int., 2014; 13: 10.1155/2014/512497
- Lynd LR, Laser MS, Bransby D, Dale BE, Davison B, Hamilton R, Himmel M, Keller M, McMillan JD, Sheehan J, et al., How biotech can transform biofuels. Nat. Biotechnol., 2008; 26:169–172.
- Maki M, Leung KT, Qin W, The prospects of cellulase-producing bacteria for the bioconversion of lignocellulosic biomass. Int. J. Biol. Sci., 2009; 5: 500–516.
- Pérez-Avalos O, Sánchez-Herrera LM, Salgado LM, Ponce-Noyola T, A bifunctional endoglucanase/endoxylanase from Cellulomonas flavigena with potential use in industrial processes at different pH. Curr. Microbiol., 2008; 57: 39–44.
- Rastogi G, Muppid GL, Gurram RN, Adhikari A, Bischoff KM, Hughes SR, et al., Isolation and characterization of cellulose-degrading bacteria from the deep subsurface of the Homestake gold mine, Lead, South Dakota, USA. J. Ind. Microbiol. Biotechnol., 2009; 36: 585–598.
- Sánchez ÓJ, Cardona CA, Trends in biotechnological production of fuel ethanol from different feedstocks. Bioresour. Technol., 2008: 99:5270–5295.
- Soares FL, Jr, Melo IS, Dias AC, Andreote FD, Cellulolytic bacteria from soils in harsh environments. World J. Microbiol. Biotechnol., 2012; 28: 2195–2203.
- Teather RM, Wood PJ. Use of Congo red-polysaccharide interactions in enumeration and characterization of cellulolytic bacteria from the bovine rumen. Appl. Environ. Microbiol., 1982; 43: 777-780.
- Waeonukul R, Kyu KL, Sakka K, Ratanakhanokchai K, Isolation and characterization of a multienzyme complex (cellulosome) of the Paenibacillus curdlanolyticus B-6 grown on Avicel under aerobic conditions. J. Biosci. Bioeng., 2009; 107: 610–614.
- Zhang M, Chekan JR., Dodd D, Hong PY, Radlinski L, Revindran V, et al., Xylan utilization in human gut commensal bacteria is orchestrated by unique modular organization of polysaccharide-degrading enzymes. Proc. Natl. Acad. Sci. U.S.A., 2014; 111: E3708–E3717.
- Zhang YHP, Himmel ME, Mielenz JR, Outlook for cellulase improvement: screening and selection strategies. Biotechnol. Adv., 2006; 24: 452–481.
© The Author(s) 2017. Open Access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License which permits unrestricted use, sharing, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.