


ISSN: 0973-7510
E-ISSN: 2581-690X
The World Health Organization documented 247 million reported malaria cases worldwide resulting in 619,000 fatalities in 2021. More than 70% of these deaths are attributed to Children under five years of age and sub-Saharan Africa is the region in which the highest number of deaths occur. The Plasmodium falciparum parasite is the deadliest form of malaria, and treating falciparum infection is becoming more challenging due to the emergence of drug-resistant parasites, causing a decrease in the efficiency of antimalarial medications. Artemisinin combination therapy is now considered the gold standard for malaria treatment; however, this method is at risk due to parasites exhibiting delayed clearance to artemisinin and resistance to partner drugs such as lumefantrine, amodiaquine, mefloquine, piperaquine, and sulfadoxine/pyrimethamine. This review assessed drug targets in Plasmodium falciparum for the development of novel antimalarials. Over Eighty-five papers on malaria, Plasmodium falciparum protein targets, and protein inhibitors were gathered from Google Scholar, ProQuest, PubMed, and Science Direct, between 2012 and 2023. Only articles with comparable keywords on malaria drug targets concentrating on enzyme proteins, carrier molecules present in Plasmodium falciparum, and their inhibitors were retrieved for review, while articles within that range that did not provide definite data were excluded. Most recently, inhibitors of dihydroorotate dehydrogenase (DHODH), artefenomel (OZ439), and ferroquine have been reported and are being explored in combination with other partner medications to work against different stages of plasmodium parasite. In identifying target proteins for drug development, essentiality and vulnerability throughout the life cycle of the parasite, its druggability, and the availability of target-based assays are critical factors. The use of modern proteomics and cellular proteins from database search which assists in parasite proliferation delivers optimal information on the new generation of lead compounds. In addition, advances in in silico methods enable the identification of protein targets for drug development.
Malaria, Resistance, Drug Targets, Drug Development, Sub-Saharan Africa, Plasmodium falciparum
The World Health Organization reported more than 247 million cases of malaria globally, with 619,000 deaths.1 More than 70% of these deaths are observed among children under five years of age, with sub-Saharan Africa being the region most affected.1-3 Because Plasmodium falciparum strains are resistant to antimalarial drugs, malaria poses a significant concern to public health, prompting development of newer scientific methodologies to enhance therapy.2,4,5 The primary genomic expression of live Plasmodium falciparum parasites is controlled by epigenetic processes and shows a specific pattern of histone alterations associated with virulence in erythrocytes.6 Currently, one of the therapies suggested by the WHO is artemisinin combination therapy, combining the medications Artemether-Lumefantrine (AL), Artesunate-Amodiaquine (AS-AQ), Artesunate-Mefloquine (AS-MQ), Artesunate-Pyronaridine (AS-PYR), Artesunate-Sulfadoxine/Pyrimethamine (AS-SP), Dihydroartemisinin-Piperaquine (DHA-PPQ). However, pharmacological resistance to quinoline analogs and artemisinin resistance raise issues regarding the safety of this treatment.7
Research has shown that proteins of Plasmodium falciparum are identified as possible drug targets for the development of antimalarial drug inhibitors.8 As a result, one of the most essential tools in drug design is understanding the relevance and susceptibility of a certain protein in the life cycles of different parasites.9
Developmental stages of Plasmodium falciparum and areas of intervention
The developmental stages of human malaria parasites are complex, but this cycle has three primary stages such; hepatic, asexual, and sexual stage. After an infected mosquito bite, sporozoites are released into the bloodstream and transported to liver cells (30-60 minutes), followed by proliferation.10
After 1-2 weeks, the sporozoites form schizonts in which more than 30,000 merozoites are housed. These schizonts within the hepatocytes releases merozoites into the bloodstream.10,11 According to Walker, Nadjm, and Whitty,12 certain sporozoites from Plasmodium ovale and Plasmodium vivax species can develop into hypnozoites. The asexual cycle then begins when merozoites infiltrate red blood cells and digest hemoglobin to develop (Figure 1). The parasite develops within the host red blood cells from the early ring stage through the late trophozoite stage, progressing to the schizont stage, which comprises of 6 to 32 merozoites.10,11
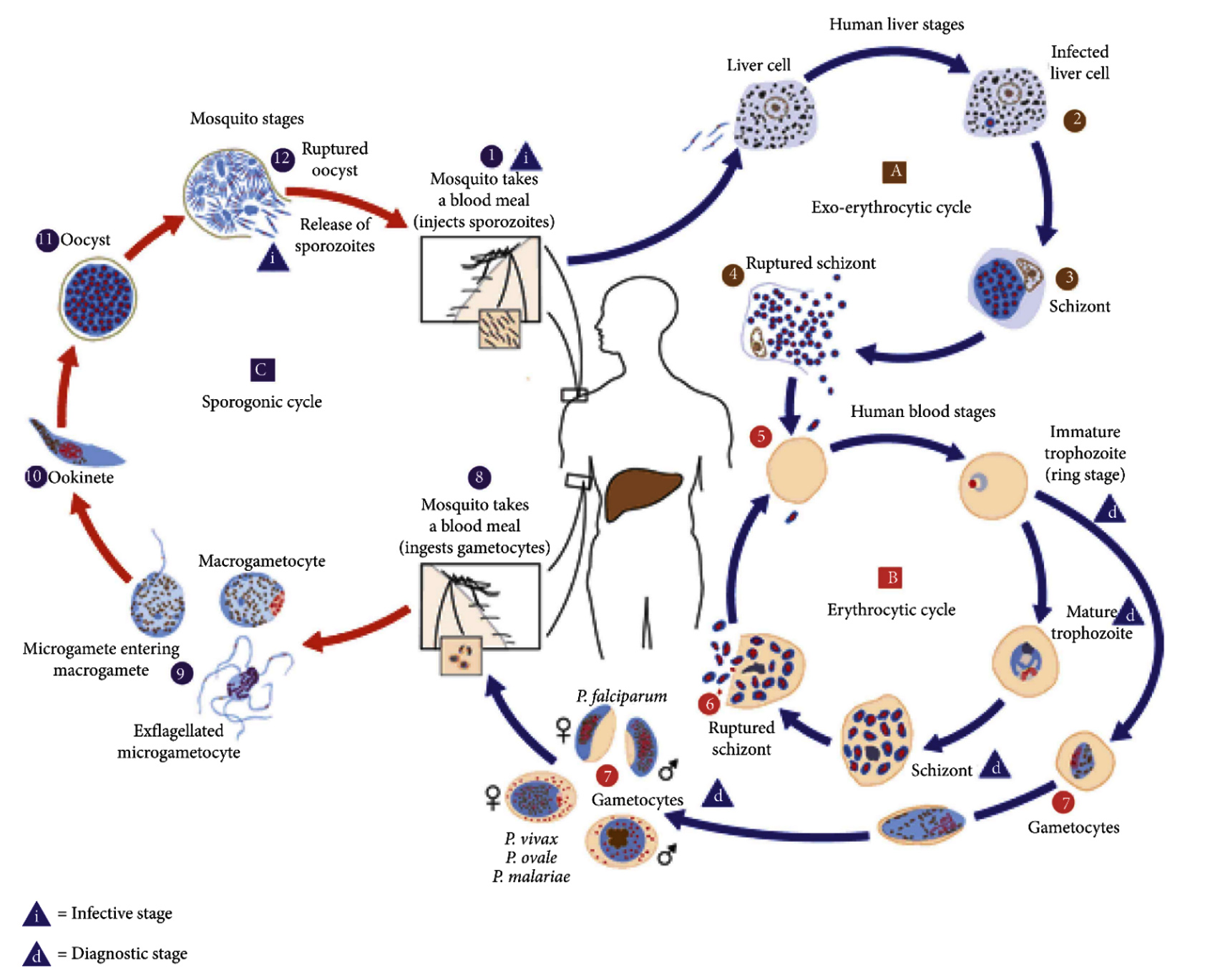
Figure 1. The parasitic cycle of Plasmodium falciparum11
In the life cycle of Plasmodium falciparum, schizonts rupture red blood cells, releasing merozoites that invade new RBCs and continue the cycle, leading to malaria symptoms. Cyclical fevers often occur soon before or after RBC lysis, when the schizonts rupture and release fresh pathogenic merozoites. Some merozoites grow throughout this cycle into erythrocytic gametocytes, These are individual male and female sexual forms with a single nucleus that can be ingested by a blood-seeking female Anopheles mosquito.10,13
The mosquito then ingests gametocytes to induce gametogenesis. The macrogametes are pierced or fertilized by Ex-flagellated microgametes, forming zygotes that develop into ookinetes and eventually into spherical oocysts. The enlargement of the oocyst and the production of multiple sporozoites are triggered by the nucleus budding off repeatedly inside the oocyst. The oocyst does bursts to release sporozoites when there are fully developed, discharging them into the hemocoel (the internal cavity of the mosquito). After reaching the glands responsible for salivary production, the sporozoites complete the cycle. The life cycle continues with the introduction of sporozoites into the bloodstream of a new human host from the salivary glands of a female anopheles mosquito during a blood meal.14
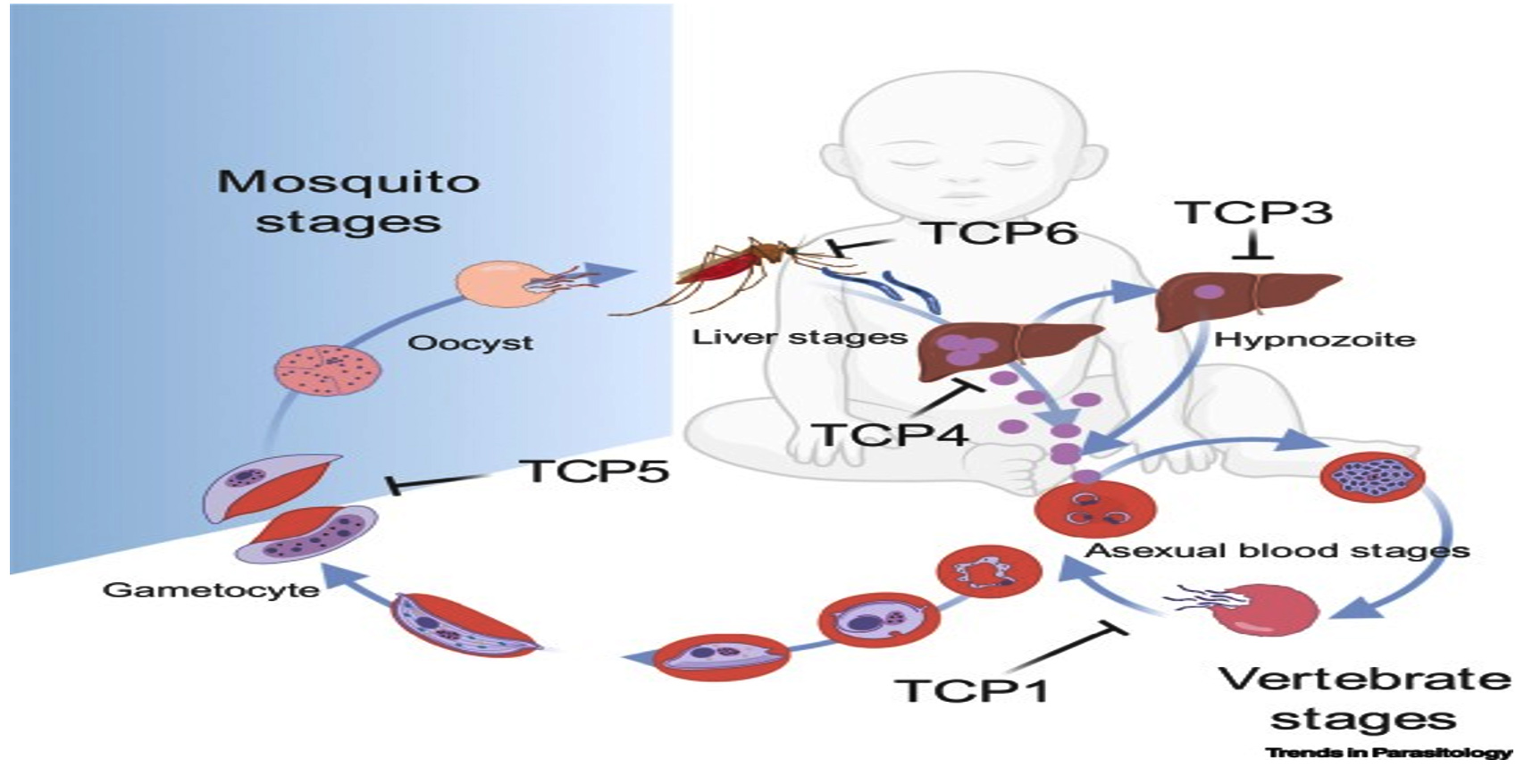
Figure 2. Plasmodium falciparum life cycle as a malaria parasite and locations of intervention15
TCPs, also known as target candidate profiles, were promoted as strategic instruments to guide the development of antimalarial therapeutic molecules in 2013 (Figure 2). Although the pathogenic phases of asexual blood are the stage that many antimalarial medications target, ideally future antimalarial treatments should also focus on other stages.15
- The five known TCPs are major in the following phases, namely:
- TCP1: compounds that eliminate parasitemia during the blood stage of asexual malaria infections.
- TCP2: compounds that can sustain antiparasitic activity.16
- TCP3: compounds inhibiting action of hypnozoites.
- TCP4: compounds that inhibit the action of hepatic schizonts.
- TCP5: compounds that prevents transmission of gametocytes.
- TCP6: mosquitocidals compounds.
However, the TCP2 molecule integrates with TCP1, it works as a long-term partner inhibitor to complete parasite eradication in the blood stage.15,17
The Google Scholar, ProQuest, and PubMed databases were searched for published peer-reviewed articles on Plasmodium falciparum drug targets that were published during the previous years between 2012 and 2023. Research articles focused on malaria drug targets, in particular enzymes and proteins, carrier molecules prevalent in Plasmodium falciparum, were investigated. In addition, documented inhibitors of these selected targets were also considered for investigation. Articles featuring Plasmodium falciparum without considerable information on therapeutic targets/proteins were excluded from the research.
Druggable Targets, Key Proteins, and their inhibitory molecule
Various proteins that indicate drug resistance and metabolic features are discovered in each of the organelles of the parasite, and these proteins span several stages of development. Due to Plasmodium falciparum’s multidrug resistance, more current drugs that function as inhibitors in various clinical trials help address the many targets in the parasite’s organelle across all of its metabolic phases.8
Pharmaceutical research has spent the last several decades exploring the diverse metabolic and biochemical processes of the parasite with the aim of discovering and applying novel treatment approaches. Research is underway to develop novel chemical compounds or specific inhibitors to target these newly identified protein targets.18
Targeting the food vacuole
The food vacuoles of falciparum malaria emerged as a potential therapeutic focus due to their vital function in sustaining parasite survival. These vacuoles serve as the main site for hemoglobin digestion, providing critical cellular materials for the growth and reproduction of the parasite within the hosts red blood cells. Researchers have identified different enzymes and proteins involved in this process, making them possible targets for antimalarial drugs.18 Inhibition of the activity of the feeding vacuole affects the acquisition of nutrients from parasites and their metabolic activities, resulting in their eventual cell death. Compounds that preferentially interfere with enzyme activity, such as plasmepsins and falcipains, which play an important role in hemoglobin degradation within food vacuoles, have shown promise in preclinical trials (Table). Targeting food vacuoles gives a tremendous opportunity to produce innovative and more effective antimalarial drugs with lower side effects, answering the urgent need for alternative therapies against drug-resistant strains of Plasmodium falciparum.19,20
Table:
Druggable targets and their respective inhibitors as addressed in this study
Drug Targets |
Function |
Enzymes, ions or carrier molecules involved |
Antimalarials in Use |
Promising Inhibitory molecules |
Mode of Action |
TCPs Targeted |
Ref. |
|---|---|---|---|---|---|---|---|
Food vacuole |
Site for heamoglobin digestion |
plasmepsins and falcipains |
ACTs Quinoline |
WM382 Quinoline derivatives |
Inhibits protease enzymes and haemozoin crystal formation |
1, 2, 4 and 6 |
[22, 25] |
PfCRT and PfMDR1 transport proteins |
Nutrient supply and metabolic waste removal |
PfCRT and PfMDR1 |
Choloroquine |
Ferroquine |
Inhibits chloroquine resistant parasite |
1 and 4 |
[29] |
P-type ATPase |
Ion transport and maintenance of cellular homeostasis |
Na+, K+, H+ Ca2+-ATPases |
KAE609, KAF156 |
SJ733 |
Targets the erythrocytic stages |
1 and 4 |
[33] |
Protein Kinases |
Proliferation and differentiation |
PfCDPK1 and PfCDPK4 |
Imidazopyridine derivatives |
KAE609 |
Interferes with proliferative and reproductive stages of the parasite |
1, 4, 5 and 6 |
[8] |
Folate pathway |
DNA synthesis |
DHPS DHFR |
Sulphadoxine Pyrimethamine, proguanil |
Sulfanilamide analogues WR99210 and P218 |
Inhibits synthesis of folic acids |
1 and 4 |
[37] |
Apicoplasts |
Protein trafficking |
HSP70chaperone-transit peptide |
15-Deoxyspergualin (DSG) |
Compound 15-Deoxyspergualin (DSG) Doxycycline |
Inhibits trafficking of proteins to apicoplasts |
1 and 4 |
[41, 8, 42] |
Mitochondria |
Electron transport and protein synthesis |
Cytochrome b1 DHODH |
Atovaquone |
Quinolones DSM265 |
Inhibits de novo pyrimidine biosynthetic pathway |
1 and 4 |
[18, 44] |
Inhibition of protease enzymes
Amino acids are essential components in the synthesis of newer proteins. These proteins are broken down by proteases, regulatory and catalytic molecules. Proteolysis, the process of breaking down proteins into smaller peptides and amino acids is facilitated by four aspartic proteases, which includes aspartic proteases (plasmepsins I, II, IV and histo-aspartic protease (HAP)), cysteine proteases (falcipains-2, falcipains-3), a metalloprotease (falcilysin), and an aminopeptidase dipeptidyl aminopeptidase 1 (DPAP1).20 Plasmoproteins and falcipanes are the main proteases involved in hemoglobin degradation.19,20 Artemisinin combination therapy (ACT) serves as a primary treatment of uncomplicated Plasmodium falciparum malaria infection by causing oxidative damage to proteins and other cellular components. This damage can affect various cellular processes, including those involved in proteases.1
Plasmodium falciparum contains plasmepsin protease ranging from I-X. Previous investigations have indicated that plasmepsin V (PMV) plays a vital role in transferring plasmodium proteins to host red blood cells, therefore triggering immunological responses.21 According to the tests by Favuzza et al.,22 WM382 serves as a promising molecule that suppresses liver, blood, and mosquito stages of the liver, blood, and mosquito stages of Plasmodium falciparum. This experimental drug has a synergistic enzymatic and inhibitory impact on plasmepsin (IX and X). Despite the redundancy in the role of food vacuole in Plasmodium and other proteases, blocking the digestive vacuole plasmepsin could be a good target for early therapeutic development.23
In Plasmodium falciparum, the primary hemoglobinase in the food vacuole are falcipains (FP1, FP2, and FP3), which are critical for digesting host hemoglobin into peptides and amino acids. Quinoline-based antimalarial drugs such as chloroquine and quinine, inhibit this hemoglobin digestion process. As a result, hemoglobin accumulates in the parasite’s food vacuole. This build up of free hemoglobin leads to increased oxidative stress within the parasite, disrupting cellular functions and ultimately causing parasite death.24
Inhibition of the production of hemozoin crystals
Malaria parasites destroy the hemoglobin present in red blood cells to consume essential amino acids, resulting in the creation of free heme, a by-product that disintegrates its cell membrane resulting from peroxidative qualities. Plasmodium falciparum oxidizes heme to hematin and then histidine-rich protein (HRP-2) facilitates the conversion of heme to hemozoin, making all of these processes druggable targets for antimalarials.25
Blood schizonticidal substances such as chloroquine, mefloquine, halofantrine, and other quinoline analogues are recognized as possible pharmacological inhibitors against hematin conversion (Figure 3). Some investigations indicated that artemisinin or its derivatives binds to heme to trigger damage to parasite structures and eventually leading to cell death.26
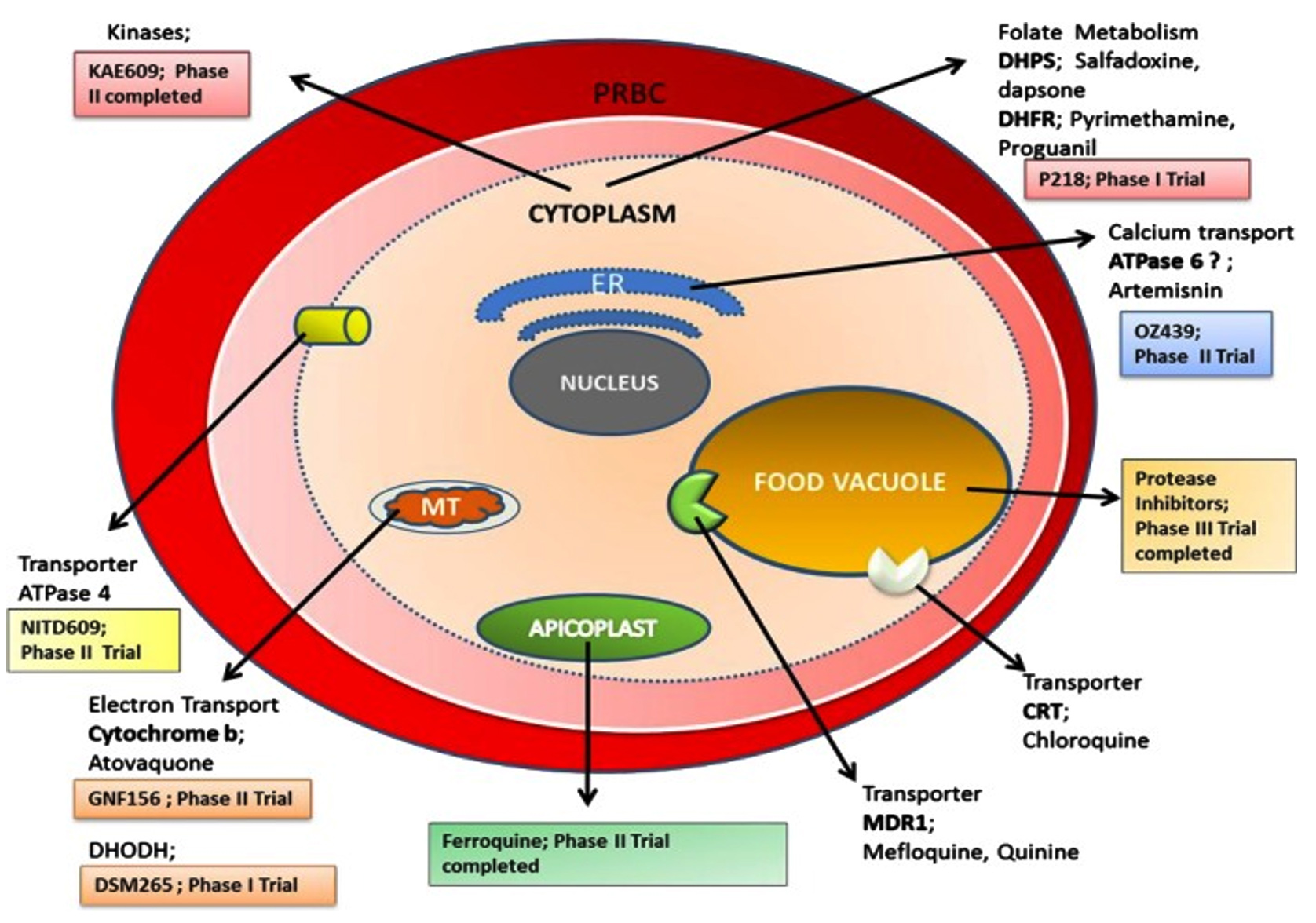
Figure 3. An overview of various druggable targets in malaria therapy8
Transport proteins of PfCRT and PfMDR1: Insight into drug resistance and targeting strategies
The Plasmodium falciparum chloroquine resistant transporter (PfCRT) and the Plasmodium falciparum multidrug resistant transporter (PfMDR1) are promising targets resulting from their important regulatory roles, ranging from their functions in utilizing essential nutrients to the removal of toxic waste in the malaria parasite.27 Both the PfCRT and PfMDR1 transport proteins are encoded by the Pfcrt and Pfmdr1 gene.28 Three main mechanisms are thought to be involved in transporter-mediated drug resistance: (i) A mutation decreasing drug effectiveness and binding affinity when the transporter is targeted; (ii) A mutation reducing drug levels at the target when the transporter acts as a delivery mechanism; and (iii) A mutation enabling drug efflux pathways, thereby decreasing effective drug concentrations at the target. The efflux transporters at the site, such as PfCRT, are known to be involved in drug resistance.18
Blood-schizonticidal ferroquine, which just passed Phase II clinical trials after it was shown to be effective against chloroquine-resistant Plasmodium falciparum, is a viable option. Unlike chloroquine, the basic structure of the 4-aminoquinoline scaffold of ferroquine allows it to accumulate in the acidic environment of the parasite’s digesting vacuole. The drug provides an alternative approach to inhibiting the chloroquine-resistant parasite in contrast to chloroquine, as an iron atom shifts between two aromatic rings to form deadly free radicals.29,30
Targeting P- type ATPase
PfATP4 is a critical P-type ATPase found in the plasma membrane of Plasmodium falciparum, the malaria-causing parasite. This enzyme plays a vital role in maintaining ionic balance within the parasite by regulating the transport of sodium (Na+) and potassium (K+) across the membrane. The activity of PfATP4 is essential for the parasite’s survival and adaptation to the hostile environment within the host. Inhibiting PfATP4 disrupts this ion homeostasis, leading to a buildup of intracellular sodium and potassium imbalances, which ultimately impairs parasite growth and function. Due to its central role in the parasite’s physiology, PfATP4 has emerged as a key target for novel antimalarial drugs designed to interfere with its ATPase activity and combat malaria more effectively.18 Notable instances of P-type ATPase encompass the sodium-potassium pump (Na+, K+ -ATPase), plasma membrane proton pump (H+ -ATPase), proton-potassium pump (H+, K+-ATPase), and calcium pump (Ca2+-ATPase). Mutations in PfATP4 can alter the efficacy of compounds such as KAE609 and SJ733 by modifying the protein’s structure and function, thereby affecting drug binding and inhibition. These mutations can lead to conformational changes in PfATP4 that reduce the affinity of these drugs for their target, thereby impairing their ability to inhibit the ATPase activity of the protein. As a result, the effectiveness of these antimalarial agents can be diminished, highlighting the importance of monitoring PfATP4 mutations to anticipate and manage potential drug resistance. Research has shown that spiroindolones [KAE609], pyrazole amides [KAF156] and dihydroisoquinolones [SJ733] are the three separate groups of effective antimalarial drugs.18,31 Through inhibition of this important protein, scientists hope to alter the parasite’s capacity to sustain its internal habitat, ultimately ending in its death. This technology offers the potential to produce novel antimalarial medications that have greater specificity and less impact on the host, thus supporting international efforts to manage and eradicate malaria. According to research by White et al.,32 KAE609 demonstrated a sevenfold increase in effectiveness compared to artesunate and was 40 times more potent than 4-aminoquinoloines. Additionally, SJ733 has undergone clinical investigation due to its favorable safety profile, oral availability, and preclinical pharmacokinetic attributes.33-35
Artefenomel (OZ439), a synthetic trioxolane, exhibits improved pharmacokinetic properties compared to current antimalarial medications containing the artemisinin pharmacophore. It is a promising candidate for targeting ATPase proteins. During its clinical stage, artefenomel has shown general tolerance in volunteers at doses up to 1600 mg. Currently, it is under investigation as a partner drug in combination therapy for malaria.36 With an in vitro efficacy equivalent to that of artemisinin derivatives used in clinical settings, artefenomel is a completely novel compound active against of all phases of malaria-causing erythrocytic asexual Plasmodium falciparum.36
Inhibition of Plasmodium falciparum protein kinases
Kinases are promising yet underexplored drug targets in malaria compared to human kinases. The extensive literature on human kinase inhibitors provides a foundation for antimalarial kinase inhibitor design, although achieving selectivity over human kinases remains challenging. Integrating target-based drug discovery with phenotypic approaches is essential for early validation. Kinase-focused phenotypic screening, chemoproteomics, and genome editing technologies enhance target identification and validation. Developing in vitro Plasmodium kinase assays and obtaining structural information will facilitate drug discovery, while non-ATP competitive inhibitors may overcome selectivity challenges. Monitoring resistance and considering polypharmacology are critical, given the high rates of resistance and structural similarities among kinases. The development of kinase-specific chemoproteomic approaches and advances in genome editing for Plasmodium provide powerful tools for target identification and validation. Plasmodium kinases, despite sharing conserved ATP sites with human kinases, may have unique features allowing selective inhibition. It is challenging to identify a superior class of kinases for antimalarial development due to potential off-target effects and the conserved nature of ATP sites. Nonetheless, Plasmodium kinases merit further investigation due to their significant therapeutic potential and the possibility of overcoming challenges related to selectivity and resistance.8,29,34-36
Targeting the folate pathway
Since the route that leads to folate metabolism supplies the essential folate cofactor for DNA synthesis, it is one of the most critical and thus a viable target for identifying antimalarial agents. Out of the seven enzymes implicated in the de novo folate synthesis process, only two have undergone thorough investigation regarding their role in malaria prevention and chemotherapy namely; dihydropteroate synthase (DHPS) and dihydrofolate reductase (DHFR). Since these enzymes are not found in humans, they could be intriguing topics for subsequent investigation.18,37
DHPS is very essential in dihydrofolate synthesis. Sulfanilamide, an inhibitor of DHPS, and the active component are produced in vivo from protosil. Subsequently, sulfanilamide analogues were created, culminating in the family of sulfur-based antifolate medications.
In both Plasmodium falciparum and other protozoa, dihydrofolate reductase functions as a bifunctional enzyme alongside thymidylate synthase (TS). However, resistance often arises due to mutations in the enzyme. Some of the most potent antimalarial medications, such as pyrimethamine, cycloguanil, as well as more recent structure-based drug design compounds WR99210 and P218, work by inhibiting the activity of the enzyme dihydrofolate reductase (DHFR).37,38
For many years, the combination of pyrimethamine and sulfadoxine medications was an excellent approach to treating malaria patients. These drugs work together to alter two separate proteins in the folate pathway. While sulfadoxine targets DHPS, pyrimethamine compounds target the DHFR enzyme. Malaria parasites acquire resistance to antifolate therapy due to changes in the dhfr and dhps genes, which actually reduces the affinity of drugs for binding to certain target enzymes.37-39
Targeting Apicoplasts involved in protein transport
Essential for the malaria parasite’s survival, the apicoplast acts as a non-photosynthetic organelle housing vital metabolic pathways for Plasmodium falciparum. These pathways, such as fatty acid synthesis, isoprenoid precursor production, and heme synthesis, are indispensable for the parasite’s survival.40 Interestingly, these important metabolic channels are lacking in the human host, but are present in different species such as bacteria, plants, and apicomplexan parasites. Hence, these parasite-specific metabolic pathways emerge as a hopeful target for developing antimalarials, given their distinct nature capable of disrupting essential functions of malaria parasites. The transportation of proteins to the apicoplast involves a dual mechanism. Initially, proteins enter the endoplasmic reticulum lumen through a hydrophobic N-terminal sequence. Subsequently, this sequence is cleaved by peptidase, facilitating protein trafficking to the apicoplast. Additionally, the process is aided by the plant plastid transit peptide. Compound 15-Deoxyspergualin (DSG), are known to exhibit antimalarial activity, restricting protein trafficking into the apicoplast by interfering with contacts with the HSP70 chaperone transition peptide.41 Antibiotics such as doxycycline target the apicoplasts in Plasmodium falciparum by inhibiting protein synthesis. Doxycycline disrupts the function of the apicoplast’s 70S ribosome, which is essential for producing proteins required for the organelle’s maintenance and function. This inhibition leads to the gradual loss of apicoplast function, ultimately resulting in the parasite’s death. Other antibiotics, like clindamycin and azithromycin, also interfere with protein synthesis or transport within the apicoplast, providing a unique mechanism of action that complements conventional antimalarial therapies and helps address drug resistance issues. These antibiotics impair the parasite’s metabolic processes by targeting the apicoplast’s protein transport pathways, leading to its elimination.42
Targeting the Mitochondria (Electron Transport Chain)
Mitochondria are primarily involved in two fundamental functions: electron transport and protein synthesis. Several antimalarial drugs, which are clinically relevant, have a confirmed effect on direct or indirect metabolism of pyrimidine. Drugs that particularly act on dihydrofolate reductase (DHFR) or dihydropteroate synthase, such as cycloguanil, pyrimethamine, sulfones, and sulfonamides, block the folate pathway, which is important for thymidine production. However, interference of atovaquone with pyrimidine metabolism leads to toxicity due to its direct targeting of the bc1 electron transport complex in mitochondria.43 A recent study clearly established that the activity of the bc1 complex is crucial to providing DHODH with oxidized ubiquinone to generate pyrimidines. Thymidylate synthase inhibitors have shown efficacy as antimalarials; although they are still in the early stages of clinical testing.
Moreover, by focusing on the electron transport chain in mitochondria, the inhibition of dihydroorotate dehydrogenase (DHODH) is achieved. DHODH, an enzyme reliant on Flavin mononucleotide, facilitates the conversion of dihydroorotate to orotate in the de novo pyrimidine biosynthesis pathway.43,44 The dihydroorotate dehydrogenase inhibitor DSM265 has just entered a phase II clinical study. It works against the liver stage and showed a favorable safety profile in phase I.18
Synergistic effects of antimalarial compounds
The synergistic effects of combining multiple antimalarial drugs have drawn substantial attention in the fight against malaria, aiming to increase the efficacy of therapy and overcome drug resistance. A popular technique includes the combination of medications with various modes of action, providing a synergistic impact that targets the parasitic protozoan at its various developmental stages. For example, the combination of artemisinin-based medications, which rapidly remove the parasite from circulation, with longer-acting treatments such as mefloquine or piperaquine, helps eradicate remaining parasites and limit resistance development.45 Synergy can also arise through the combination of medications that target various biochemical pathways within the parasite, for instance, combining chloroquine, which disrupts heme detoxification in parasite digestive vacuoles, with atovaquone, which hampers the electron transport chain in mitochondria, has demonstrated synergistic outcomes. Such combinations not only boost the overall antimalarial activity but also minimize the risk that the parasite will acquire resistance to the medications.
In addition, research on combination treatments, which involves the use of various medications with various mechanisms of action, is gaining traction. This technique seeks to maximize therapeutic efficacy, reduce resistance, and reduce the duration of therapy. However, clinical trials for malaria therapy have progressed through phase I and phase II for dihydroorotate dehydrogenase (DHODH) inhibitors Artefenomel (OZ439) and Ferroquine (FQ), which have been published and are being tested in combination with other partner drugs to act against different stages of the parasite. Examples of these combinations are Artefenomel with piperaquine, ferroquine-artesunate, and Artefenomel-ferroquine.46 According to previously published studies, ferroquine with artesunate combination exhibits high selective toxicity against the parasite and its safe in all doses studied, having good curative rates against Plasmodium falciparum malaria. This combination of medications shows promise for the treatment of the condition. Furthermore, ferroquine can be combined with other partner drugs to produce a newer class of antimalarial combinations, particularly in places where artemisinin combination treatment (ACT) has been less effective.47 Another benefit of ferroquine with artesunate over other partner drugs in lumefantrine-containing ACTs is that patients can get this combination once daily for three days rather than twice, increasing patient adherence to therapy.46,47 Ongoing research on the identification and improvement of synergistic drug combinations remains vital to combating this chronic and fatal disease.
Future perspectives
The future of antimalarial drug therapy is promising, as researchers continue to investigate creative techniques to overcome drug resistance, increase treatment efficacy, and limit adverse effects. An area of investigation involves the creation of new medications with unique mode of action that interfere with different developmental phases of the malaria parasite. Advances in genomics, proteomics, and high-throughput screening techniques are easing the discovery of novel therapeutic targets, enabling the formulation of drugs that are not only effective against strains, but also less prone to resistance development.30
New deadly target proteins from proteomic research, in silico parasite studies, and new antimalarial drugs are necessary, since parasite resistance compromised the efficacy of presently existing antimalarial drugs. This updated technique will facilitate the identification of target proteins for a new antimalarial drug that is safer and more effective.
Limitations to antimalarial drug discovery
The discovery of antimalaria drugs faces various obstacles that hinder the evolution of effective therapies to combat falciparum malaria. The emergence and transmission of drug-resistant strains of falciparum malaria present a significant challenge. Malaria parasites demonstrate a notable capacity to develop resistance against commonly prescribed antimalarial drugs, including chloroquine and artemisinin-based combination treatments (ACT).45 Resistance affects the efficacy of established therapies and underlines the urgent need for new therapeutic methods.
Furthermore, the complicated stages of the parasites life forms hampers the development of medications, as the malaria parasite traverses many phases in both the insect and blood stage, each with specific biological properties. Targeting the parasite at different phases requires a deep understanding of its biology, which adds complexity to drug research attempts. Insufficient in vitro and in vivo models that correctly mimic all phases of the malaria life cycle further impedes the discovery and evaluation of viable treatment candidates.48
Additionally, inadequate funding for malaria research and treatment development represents a substantial limitation. Malaria largely affects populations in resource-limited locations, and as a result, there is a lack of financial incentives for pharmaceutical companies to invest in substantial research and development for novel antimalarial drugs.49 This budgetary limitation hinders the development of new drug candidates and hinders progress in identifying more effective and cheaper therapies for malaria. Addressing these constraints requires a coordinated effort from the global health community to prioritize and support research activities to overcome hurdles in the creation of antimalarial drugs.
Discovering inhibitors that effectively target essential proteins and cellular processes of Plasmodium falciparum across its life cycle is crucial for producing new antimalarial drugs. A comprehensive approach emphasizes the importance of sensitivity, necessity, and feasibility of target-based assays in vitro research. Successful suppression of these pharmacological targets forms the foundation for developing effective antimalarial medications, offering promising prospects in the ongoing battle against malaria. Encouraging collaborative endeavors, interdisciplinary investigations, and the exploration of cutting-edge technologies can expedite the identification and development of potent antimalarial drugs that specifically target key proteins in Plasmodium falciparum, thereby curbing its transmission among populations.
ACKNOWLEDGMENTS
The authors would like to thank Management of Covenant University Centre for Research Innovation and Discovery (CUCRID) for defraying the necessary APC cost and also acknowledge the Department of Biological Sciences, College of Science and Technology, Covenant University, Nigeria, for their support.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
AUTHORS’ CONTRIBUTION
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
FUNDING
This study was funded by Covenant Applied Informatics and Communication Africa Center of Excellence (CApIC-ACE), Scholarship, Covenant University, Ota, Nigeria.
DATA AVAILABILITY
All datasets generated or analyzed during this study are included in the manuscript.
ETHICS STATEMENT
Not applicable.
- World Health Organization. World malaria report 2022. 2022. https://www.who.int/teams/global-malaria-programme/reports/world-malaria-report-2022 [Accessed: 26 April 2023].
- De Sousa ACC, Combrinck JM, Maepa K, Egan TJ. Virtual screening as a technique to uncover novel b-haematin inhibitors with effectiveness against malaria parasites. Sci Rep. 2020;10(1):3374.
Crossref - Adegbite G, Edeki S, Isewon I, et al. Mathematical modeling of malaria transmission dynamics in humans with mobility and control states. Infect Dis Model. 2023;8(4):1015-1031.
Crossref - Olasehinde GI, Diji-geske RI, Fadina I, et al. Epidemiology of Plasmodium falciparum infection and drug resistance markers in Ota Area, Southwestern Nigeria. Infect Drug Resist. 2019;12:1941-1949.
Crossref - Duffy S, Avery VM. Identification of inhibitors of Plasmodium falciparum gametocyte development. Malaria J. 2013;12(1):408.
Crossref - Zhao R, Wang H-H, Gao J, et al. Plant volatile compound methyl benzoate is highly effective against Spodoptera frugiperda and safe to non-target organisms as an eco-friendly botanical-insecticide. Ecotoxicol Environ Saf. 2022;245(114101):114101.
Crossref - World Health Organization. 2018. World malaria report 2017. Geneve, Switzerland: World Health Organization. [Accessed: 18 March 2023].
- Sinha S, Medhi B, Sehgal R. Challenges of drug-resistant malaria. Parasite. 2014;21:61.
Crossref - Arendse LB, Wyllie S, Chibale K, Gilbert IH. Plasmodium kinases as possible therapeutic targets for malaria: Challenges and prospects. ACS Infect Dis. 2021;7(3):518-534.
Crossref - Nureye D, Assefa S. Old and current achievements in life cycle, pathophysiology, diagnosis, prevention, and treatment of malaria including views in Ethiopia. Sci World J. 2020;2020(10):1295381.
Crossref - Bhowmik B, Chiranjib B, Singh N, Jaiswal K, Kumar KP. Recent advancements in prevention, treatment and medicine of malaria. J Chem Pharm Res. 2010;1(2):83-90.
- Walker NF, Nadjm B, Whitty CJM. Malaria. Medicine. 2010;38 (1): 41-46.
Crossref - Adedeji EO, Ogunlana OO, Fatumo S, et al. Anopheles metabolic proteins in malaria transmission, prevention and control: a review. Parasit Vectors. 2020;13(1):465.
Crossref - CDC. The History of Malaria, an Ancient Disease, Division of Parasitic Diseases and Malaria, USA. 2016. [Accessed on 17 December 2022].
- Yang T, Ottilie S, Istvan ES, et al. MalDA, speeding malaria drug discovery. Trends Parasitol. 2021;37(6):493-507.
Crossref - Umumararungu T, Nkuranga JB, Habarurema G, et al. Recent developments in antimalarial drug discovery. Bioorg Med Chem. 2023;88-89(117339):117339.
Crossref - Burrows JN, van Huijsduijnen RH, Mohrle JJ, Oeuvray C, Wells TNC. Designing the next generation of drugs for malaria control and eradication. Malaria J. 2013;12(1):187.
Crossref - Kumar S, Bhardwaj TR, Prasad DN, Singh RK. Drug targets for resistant malaria: Historic to future perspectives. Biomed Pharmacother. 2018;104:8-27.
Crossref - Alam A. Serine proteases of malaria parasite Plasmodium falciparum: Potential as antimalarial drug targets. Interdiscip Perspect Infect Dis. 2014;2014:453186.
Crossref - Abugri J, Ayariga J, Sunwiale SS, et al. Targeting the Plasmodium falciparum proteome and organelles for potential antimalarial drug candidates. Heliyon. 2022;8(8):e10390.
Crossref - Mathews ES, Odom JAR. Tackling resistance: emerging antimalarials and new parasite targets in the era of elimination. F1000Research. 2018;7:1170.
Crossref - Favuzza P, de Lera Ruiz M, Thompson JK, et al. Dual plasmepsin-targeting antimalarial drugs disrupt various phases of the malaria parasite life cycle. Cell Host Microbe. 2020;27(4):642-658.
Crossref - Belete TM. Recent progress in the development of new antimalarial drugs with novel targets. Drug Des Devel Ther. 2020;14:3875-3889.
Crossref - Combrinck JM, Mabotha TE, Ncokazi KK, et al. Insights on the involvement of heme in the mechanism of action of antimalarials. ACS Chem Biol. 2013;8(1):133-137.
Crossref - Sigala PA, Goldberg DE. The peculiarities and paradoxes of Plasmodium heme metabolism. Ann Rev Microbiol. 2014;68(1):259-278.
Crossref - Wang J, Zhang C-J, Chia WN, et al. Haem-activated promiscuous targeting of artemisinin in Plasmodium falciparum. Nat Commun. 2015;6(1).
Crossref - Ecker A, Lehane AM, Clain J, Fidock DA. PfCRT and its role in antimalarial drug resistance. Trends Parasitol. 2012;28(11):504-514.
Crossref - Pimpat Y, Saralamba N, Boonyuen U, et al. Genetic study of the orthologous crt and mdr1 genes in Plasmodium malariae from Thailand and Myanmar. Malaria J. 2020;19(18):315.
Crossref - Biamonte MA, Wanner J, Le Roch KG. Recent advances in malaria drug discovery. Bioorg Med Chem Lett. 2013;23(10):2829-2843.
Crossref - Munro BA, McMorran BJ. Antimalarial drug strategies to target Plasmodium gametocytes. Parasitologia. 2022;2(2):101-124.
Crossref - Rottmann M, McNamara C, Yeung BKS, et al. Spiroindolones, a potent compound class for the treatment of malaria. Science. (New York, N.Y.). 2010;329(5996):1175-1180.
Crossref - White NJ, Pukrittayakamee S, Phyo AP, et al. Spiroindolone KAE609 for falciparum and vivax malaria. N Eng J Med. 2014;371(5):403-410.
Crossref - Spillman NJ, Kirk K. The malaria parasite cation ATPase PfATP4 and its role in the mechanism of action of a new arsenal of antimalarial drugs. Int J Parasitol Drugs Drug Resist. 2015;5(3):149-162.
Crossref - Derbyshire ER, Zuzarte-Luis V, Magalhaes AD, et al. Chemical interrogation of the malaria kinome. Chembiochem. 2014;15(13):1920-1930.
Crossref - Ojo KK, Eastman RT, Vidadala R, et al. A specific inhibitor of PfCDPK4 blocks malaria transmission: Chemical-genetic validation. J Infect Dis. 2014;209(2):275-284.
Crossref - Chapman TM, Osborne SA, Wallace C, et al. Optimization of an imidazopyridazine series of inhibitors of Plasmodium falciparum calcium-dependent protein kinase 1 (pfCDPK1). J Pharm Chem. 2014;57(8):3570-3587.
Crossref - Yuthavong Y, Tarnchompoo B, Vilaivan T, et al. Malarial dihydrofolate reductase as a paradigm for drug development against a resistance-compromised target. Proc Natl Acad Sci U S A. 2012;109(42):16823-16828.
Crossref - Nzila A. The past, present and future of antifolates in the treatment of Plasmodium falciparum infection. J Antimicrob Chemother. 2006;57(6):1043-1054.
Crossref - Nzila A. Inhibitors of de novo folate enzymes in Plasmodium falciparum. Drug Discov Today. 2006;11(19-20):939-944.
Crossref - van Dooren GG, Striepen B. The algal past and parasite present of the apicoplast. Ann Rev Microbiol. 2013;67(1):271-289.
Crossref - Banerjee T, Singh RR, Gupta S, Surolia A, Surolia N. 15-deoxyspergualin hinders physical interaction between basic residues of transit peptide in PfENR and Hsp70-1. IUBMB Life. 2012;64(1):99-107.
Crossref - Gaillard T, Madamet M, Pradines B. Tetracyclines in malaria. Malaria J. 2015;14(1).
Crossref - Phillips AM, Rathod PK. Plasmodium dihydroorotate dehydrogenase: A promising target for novel anti-malarial chemotherapy. Infect Dis Drug Targets. 2010;10(3):226-239.
Crossref - Phillips MA, Lotharius J, Marsh K, et al. A long-duration dihydroorotate dehydrogenase inhibitor (DSM265) for prevention and treatment of malaria. Sci Transl Med. 2015;7(296):296ra111.
Crossref - Ashley EA, Dhorda M, Fairhurst RM, et al. Spread of Artemisinin Resistance in Plasmodium falciparum Malaria. N Engl J Med. 2014;371(5):411-423.
Crossref - Abd-Rahman AN, Zaloumis S, McCarthy JS, Simpson JA, Commons RJ. Scoping review of antimalarial drug candidates in phase I and II drug development. Antimicrob Agents Chemother. 2022;66(2):e0165921.
Crossref - Held J, Supan C, Salazar CLO, Tinto H, et al. Ferroquine and artesunate in African adults and children with Plasmodium falciparum malaria: a phase 2, multicentre, randomised, double-blind, dose-ranging, non-inferiority study. Lancet Infect Dis. 2015;15(12):1409-1419.
Crossref - Mishra M, Singh V, Singh S. Structural insights into key Plasmodium proteases as therapeutic drug targets. Front Microbiol. 2019;10.
Crossref - Ridley RG. Medical need, scientific opportunity and the drive for antimalarial drugs. Nature. 2002;415(6872):686-693.
Crossref
© The Author(s) 2024. Open Access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License which permits unrestricted use, sharing, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.