


ISSN: 0973-7510
E-ISSN: 2581-690X
Coxsackievirus B3 is an Enterovirus implicated in diverse human pathologies, from viral myocarditis to neurological disorders. There isn’t a medicinal agent or vaccine for CVB3 in clinical use at the moment, despite the possibility that vaccination could lower the prevalence of these illnesses. This study focuses on the in vitro production and characterization of the viral protein 1 (VP1) in the objective to use it as subunit vaccine and/or immunodiagnostic reagent. VP1 is considered as the most immunogenic capsid protein of the CVB3 surface. We amplified the VP1 whole gene by RT-PCR from the extracted wild type Nancy strain RNA, then cloned it into the pUC19 plasmid expression vector, and expressed it in E. coli DH5a prokaryotic cells. The obtained recombinant proteins were then analyzed by SDS-PAGE and characterized by Bioinformatic software tools. Our results revealed that the produced recombinant amino acid VP1 (rVP1) is highly identical to the VP1 of the CVB3 wild-type strain and has very similar physicochemical properties. In addition, we demonstrated that rVP1 has the highest number of phosphorylation sites which means that rVP1 can translate the host cell signal via the phosphorylation mechanism. Moreover, The Linear B cell epitope analysis showed that the rVP1 contains many epitope regions that should be recognized by the humoral host immune response. Taken together, results demonstrate that the cloned and recombined expressed viral protein could be used to carry out any studies concerning the development a protein subunit vaccine against CVB3 infections or an immunodiagnostic reagent for detecting the virus in samples.
CVB3, rVP1, Cloning, Molecular Characterization, Vaccine, Diagnosis
Coxsackievirus B3 (CVB3) is a non-enveloped single-stranded RNA virus and belongs to the genus Enterovirus of the Picornaviridae family1 Coxsackievirus type B3 has been linked to many human illnesses, including viral myocarditis and neurological conditions, long recognized as one of the principal causes of viral myocarditis, which can result in both acute and chronic heart failure. Furthermore, several clinical experimental and epidemiological data suggest an association between enteroviral infection, particularly CVB, and Type 1 Diabetes (T1D), which is the consequence of the destruction of b-cells in the islets of Langerhans of the pancreas. In addition, Coxsackieviruses A, B and enterovirus A71 are responsible for the acute disease known as enteroviral vesicular stomatitis hand, foot and mouth disease (HFMD). It mostly affects youngsters under the age of ten. But it might also impact adults.2,3 However, it can occasionally result in severe neurological issues such as encephalitis, meningitis, and paralysis like that brought on by the poliovirus. This variety is highly deadly and has a high fatality rate.4 About 5% of enteroviral infections result in encephalitis.5 Coxsackieviruses A and B, echoviruses6 and enterovirus A715 are the leading causes of this severe neurological disorder. The icosahedral capsid of CVB3 has a diameter of approximately 30 nm and contains the RNA genome.7,8 The RNA is 7.4 kb long and single-stranded positive-sense.9 The 5′ and 3′ untranslated regions (UTRs) define the perimeter of the CVB’s extended open reading frame (ORF). The 5′ UTR region binds covalently to the essential polar viral protein VPg. It contains an internal ribosomal entry site (IRES) (Figure 1), which is crucial in initiating mRNA translation at the ribosome level.10 The 3’UTR region mainly comprises the polyA sequence, which is probably involved in the replication of viral RNA.11 A large monocistronic polyprotein produced by the ORF is translated into one structural polyprotein (P1) and two non-structural polyproteins (P2 and P3).12 P1 is the structural capsid protein that forms the icosahedral capsid structure of the virus from VP1 to VP4, while P2 and P3 are the seven non-structural viral proteins (2A, 2B, 2C, 3A, 3B, 3C, and 3D).13 As a result of the promotion of conformational changes, neutralizing antibodies and anti-picornavirus medications can attach to the hydrophilic VP1 tunnel and prevent the virus from proliferating. Furthermore, the primary B-cell epitope in VP1 makes it the most immunogenic protein on the coxsackievirus surface.14 There is no known cure or vaccine for CVB3, even though it ranks among the most common causes of unintentional mortality in infants and young children.15 Developing a vaccine against non-poliomyelitis EV strains seems to be the only way to fight against the viral infection. Unfortunately, vaccine trials are rare, and their results are unclear. Among the most encouraging trials, we cite the immunization of mice by two successive intramuscular injections of the plasmid pCMV containing the gene encoding the capsid protein VP1 followed by a percentage of survival greater than 70%, four weeks after infection with the strain CVB3. On the other hand, a bivalent VP1 gene vaccine plasmid of CVB1 and CVB3 was obtained. The gene vaccine thus constructed induces CVB-specific antibodies in mice and can serve as a gene vaccine candidate for human beings. In addition, the injection of a plasmid coding for VP3 and VP4 followed by a viral infection gave fewer convincing results from the point of view of mouse survival time. The intensity of the cell-mediated immune response elicited by VP3 and VP4 is weaker than that induced by the capsid protein VP1. In another study, some recombinant CVB3 plasmids encode CVB3 capsid proteins (VP1 or VP3) when mice were inoculated and challenged, the death of myocytes was lessened, and the survival rates were higher than in the control group. pCA-VP3 and pCA-VP1 are suitable candidates for a CVB3 DNA vaccination. According to in vivo experiments, oral vaccination of BALB/c mice with pBBADs-VP1- transformed bacteria generated powerful immune responses against EV71 infection, including virus-neutralizing titers, anti-EV71-VP1 antibody, and the development of The immune responses in the spleen and Peyer’s patches. Importantly, neonatal mice were protected when their mothers were immunized with this recombinant Bifidobacterium longum (B. longum) that expresses VP1. These findings show that the novel oral vaccination using B. longum that expresses the VP1 protein may successfully trigger a particular immune response against EV71 infection. To stop the spread of the infection in the population, particularly newborns and the immunocompromised, vaccination against CVB3 is desirable. The future of vaccination is full of promise because of recent developments in molecular biology and genetic engineering. Starting from these data, it supposed that the use of rVP1 can develop a vaccine against CV-B3 infections and an immunodiagnostic reagent.
Viral strain, bacterial strain, Plasmid, and growing medium
We used Human Coxsackievirus B3 wild-type Nancy strain (a gift from the Virology Laboratory of the Biotechnology Department of the University of Monastir, Tunisia), the bacterial strain Escherichia coli DH5³ (SupE44, lacU169 (F80 lacZM15), hsdR17, recA1, thi-1, endA1, gyrA96 relA1), The commercial vector plasmid of the University of California (pUC19) (Invitrogen) and the bacterial strain was grown in either rich or low ampicillin (Amp; 100 µg/mL) liquid Lambda-Broth (LB).
CVB3 VP1 gene amplification by PCR
Two distinct procedures were used to extract the total RNA (viral genome): the approach based on utilizing the RNA Extraction Kit (Haven business) in accordance with the manufacturer’s instructions, and the method described by Chomczynski and Sacchi using isothiocyanate or TRI-Reagent®.16 The higher caliber extracted RNA was applied to subsequent experiments. The primers CVB3-VP1-F:
52-TTTTATTACGGTGATGGTATC-32 and CVB3-VP1-R: 52-TTTGAAGTAGATTCTAATGGT-32 were used to amplify the capsid VP1 gene of the CVB3 strain Nancy. Following a three-minute pre-denaturation at 94°C, 35 cycles of amplification were conducted, with each cycle consisting of three steps: denaturation (30 seconds at 94°C), hybridization (30 seconds at 45°C), and elongation (1 minute at 72°C). One more elongation phase was carried out for seven minutes at 72°C. The restriction sites for the VP1 genes CVB3-VP1-F’: 52 -TATCGAATTCGGGCCAACAGAGGAATCTGTGG-32 and CVB3-VP1-R’: 52-TATCGGATCCAGTGG TTACCAGACTTGCACGC-32 were added using a few more primers. EcoRI and BamHI restriction sites are represented by the sequences that are underlined, respectively. An initial denaturing step of 3 minutes at 94°C was followed by 35 cycles of denaturation at 94°C for 1 minute, annealing at 65°C for 1 minute, and extension at 72°C for 1 minute, followed by a final extension step of 10 minutes at 72°C, for the amplification of the VP1 gene limited on both sides by the restriction sites. After that, a 1% agarose gel was used to confirm the PCR results, and ethidium bromide was used to stain them. Sanger method sequencing was carried out using an AB PRISMTM 310 Genetic Analyzer (Applied Biosystems, Waltham, MD, USA).17
Cloning of the VP1 gene into expression plasmid vector
The CVB3 Nancy wild type strain’s VP1 gene sequence was cloned into the pUC19 vector, we digested the EcoRI-VP1-BamHI sequence and the cloning vector with EcoRI/BamHI restriction enzyme. Then, we carried out a ligation between the digested vector and our insert using a T4 DNA ligase. Visualization is done by migration in a 1% agarose gel electrophoresis.
Expression of the recombinant VP1 (rVP1) in prokaryotic system
The competent E. coli DH5α cells were produced using the calcium chloride method, which has been previously reported elsewhere.18 The ligation products were transformed into recombinant clones in the competent cells selected by the PCR colony. Each colony was removed from LB plates supplemented with Amp using a sterile tip, and it was then dissolved in the appropriate PCR mixture. Primers designed specifically for the inserts were used. After using the previously described PCR technique for 40 cycles of amplification, the PCR products were found using 1% agarose gel electrophoresis. Following purification of the recombinant plasmid pUC19-VP1 with a “EZ-10 Spin Column Plasmid DNA Kit” (BIO BASIC, Canada), restriction analysis and an AB PRISMTM 310 Genetic Analyser (Applied Biosystems, Waltham, MD, USA) were used to sequence the recombinant VP1 (rVP1). Based on the Sanger approach, it was digested for two hours at 37°C using 15 units of EcoRI and BamHI (TaKaRa, Canada).17
Characterization of the rVP1 subunit protein by SDS-PAGE
The procedures for separating and analyzing protein samples using SDS-PAGE were followed by Laemmli. This was done in 15% polyacrylamide gels at room temperature using the Mini-Protean tetra cell 4-gel hand casting technique (BIO-RAD, China). Ten microliters of the sample and five microliters of the pre-stained protein ladder (10-250 kD) were put into each well. The gel was stained until the gel background was clear using Coomassie Blue staining solution and distain solution (40 mL methanol combined with 7 mL glacial acetic acid and 53 mL distilled water to make a final volume of 100 mL). The gel was operated at 25 mA for around two hours. On 15% SDS-PAGE, the recombinant CVB3 Viral Protein (rVP1) was separated.
Bioinformatics analysis
The “cdd” database (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) was utilized for the examination of conserved domains, and the Clustal Omega program (https://www.ebi.ac.uk/Tools/msa/clustalo/) was utilized for sequence comparison. Next, we used the Sequence Manipulation Suite (SMS) (https://www.bioinformatics.org/sms2/translate.html) to translate the DNA sequences of rVP1 and the capsid proteins of the CVB3 Nancy strain into protein sequences. Then, we examined the physicochemical characteristics of the rVP1 and capsid proteins of CVB3 strain Nancy using the bioinformatics program ProtParam of the ExPASy service (https://web.expasy.org/protparam/). The Signal P server (https://services.healthtech.dtu.dk/services/SignalP-6.0/), the “NetPhos Server” (https://services.healthtech.dtu.dk/services/NetPhos-3.1/), and the N-glycolysis prediction sites (https://services.healthtech.dtu.dk/services/NetNGlyc-1.0/) were used to predict the presence of signal peptides.19,20 The functional domains21,22 were analyzed using InterPro (https://www.ebi.ac.uk/interpro/search/sequence/), and acetylation prediction at the internal lysine level was predicted using PAIL (http://pail.biocuckoo.org/online.php). Using SOPMA (https://npsa-prabi.ibcp.fr/cgi- bin/npsa_automat.pl?page=/NPSA/npsa_sopma.html), we were able to predict the secondary structures of the rVP1 and CVB3 strain Nancy capsid proteins. For the protein structural homology modeling service, we employed SWISS-MODEL. Predicting homologous sequences is the goal of this site (https://swissmodel.expasy.org/interactive). Three-dimensional structures can be viewed using the macromolecular structure viewer program Cn3D (https://www.ncbi.nlm.nih.gov/Structure/icn3d/). In this work, the Protein Data Bank (PDB) file type was utilized. The proteins’ β-cell epitopes were predicted using Bepipred (https://services.healthtech.dtu.dk/services/BepiPred-2.0/).
Amplification of the wild type CVB3 VP1 gene
We performed a genomic amplification by RT-PCR of the VP1 sequence (one intact and one added by the EcoRI and BamHI enzymatic digestion sites) in the wild type Nancy CVB3 strain. The amplification products revealed on a 1% agarose gel show the following result (Figure 1).
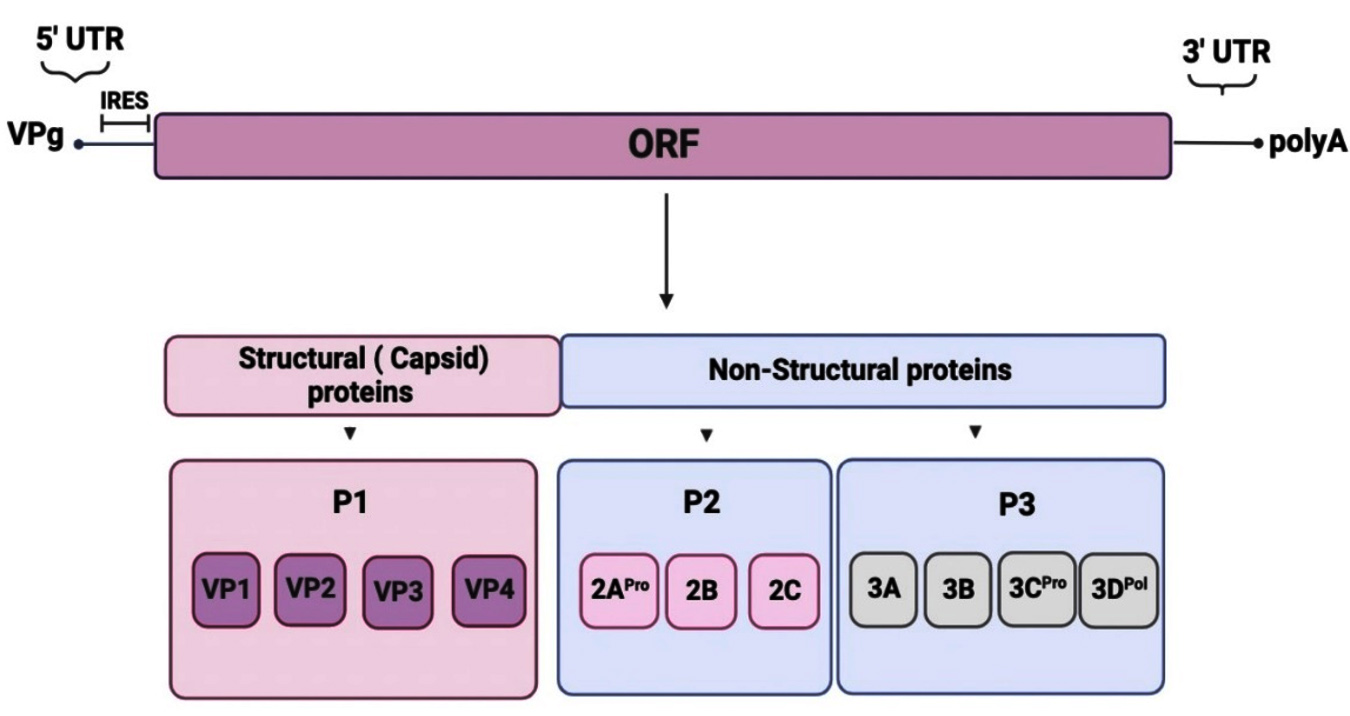
Figure 1. Genomic organization of CVB3. The genome’s highly organized untranslated region (UTR) sections are located at the 5′ and 3′ ends. The 5′-UTR contains the internal ribosome entry site (IRES) required for cap-independent translation. The genome encodes a single polyprotein with three distinct P1 to P3 regions
Cloning of the VP1 gene into the pUC19 expression vector
pUC19 is digested simultaneously by both EcoRI and BamHI restriction enzymes, giving two fragments of different sizes. The first fragment is the one that will serve in cloning after its purification from the gel. After pUC19 was digested by restriction enzymes and the VP1 sequence was amplified by EcoRI and BamHI primers specific for adding restriction sites, we performed the ligation step, which should result in one band on 1% agarose gel. To confirm the success of the ligation step, we did the digestion of recombinant pUC19 plasmid by EcoRI and BamHI restriction enzymes. After revelation on 1% agarose gel, we obtained two bands corresponding to the VP1 gene and the vector (Figure 2).
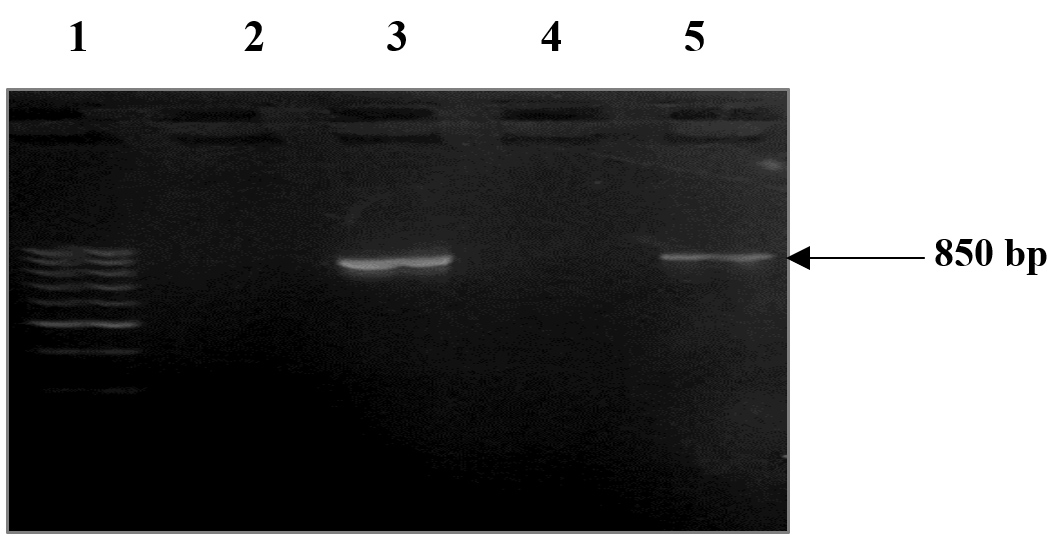
Figure 2. Revelation on 1% agarose gel of the VP1 sequence of CVB3 Nancy strain. Lane 1 corresponds to the molecular weight (MW) (50 bp -1000 bp). Lanes 2 and 4 correspond to the negative control (double distilled water). Lanes 3 and 5 correspond to the amplification product of the VP1 region of CVB3 successively with and without the addition of the EcoRI and BamHI enzymatic digestion sites. The entire nucleotide sequence of the genome of the CVB3 Nancy strain was used to design the forward and reverse primers, which were intended to amplify the VP1 gene region, which is roughly 850 bp in size. The negative control certifies the absence of contamination and the PCR amplification specificity
Bacteria transformation
Expression of the transformation vector
After the transformation of the bacteria strain E. coli DH5a by creating pores in the wall of bacteria to let the plasmids penetrate, then using a heat shock to transform the vector into these bacteria artificially, we made a spreading of this bacteria on LB medium with ampicillin. In fact, after spreading the transformation product on this medium, only the bacteria having incorporated the recombinant plasmids can grow on the medium because they contain the ampR gene, which is resistant to ampicillin, but the untransformed E. coli DH5a strain is sensitive to ampicillin therefore, they will not grow on this medium. The Figure 3 shows the different results of the transformation.
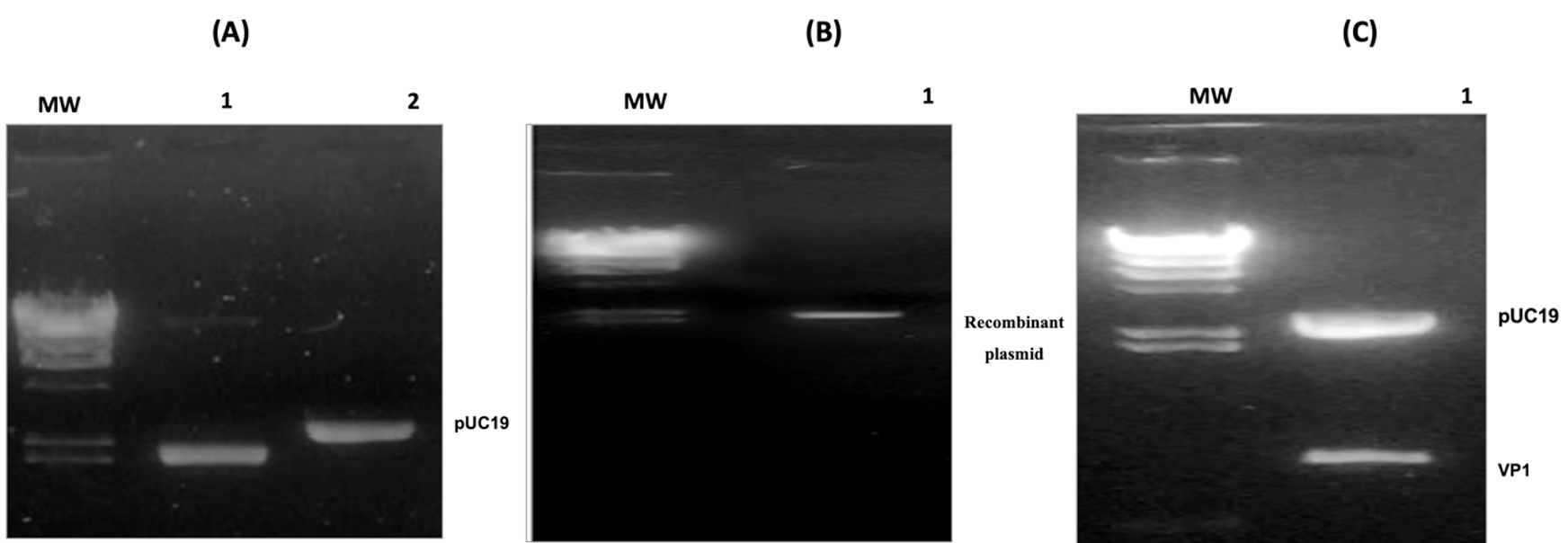
Figure 3. Confirmation of the cloning process. (A) MW corresponds to the molecular weight (Phage Lamda DNA digested by HindIII), lane 1 corresponds to PUC19 plasmid digested by enzymes and lane 2 contains the same plasmid undigested. (B) MW corresponds to the molecular weight (Phage Lamda DNA digested by HindIII) and lane 1 corresponds to the recombinant plasmid. (C) The MW Lane corresponds to the Lambda DNA/HindIII Plus size marker, and the pUC19-VP1 lane corresponds to the product of the double enzymatic digestion of the recombinant plasmid. This result shows two bands, each corresponding to the pUC19 vector (~2686 bp) and the VP1 insert (~850 bp). This confirms that the band above represents the recombinant pUC19 expression vector containing the VP1 insert
Screening colonies by PCR method and Extraction of the recombinant plasmid from the recombinant colonies
We used colony PCR to confirm that the recombinant plasmid (pUC19-VP1) was incorporated into the bacteria. Recombinant colonies can be found using this form of screening. To accomplish this, we amplified the VP1 gene sequence using the VP1-F and VP1-R primers utilized previously in PCR gene amplification. From the colonies growing on the LB medium with ampicillin. We extracted recombinant plasmids using the EZ-10 Spin column plasmid DNA kit (BioBasic, Canada). Obtained results (Figure 4) demonstrated confirm that the bands already found in the gel profile on colonies correspond to the recombinant pUC19 plasmid. Digestion of the plasmid DNA by the same enzymes used previously and the sequencing of the recombinant plasmid are necessary to confirm or invalidate the obtaining of a clone.
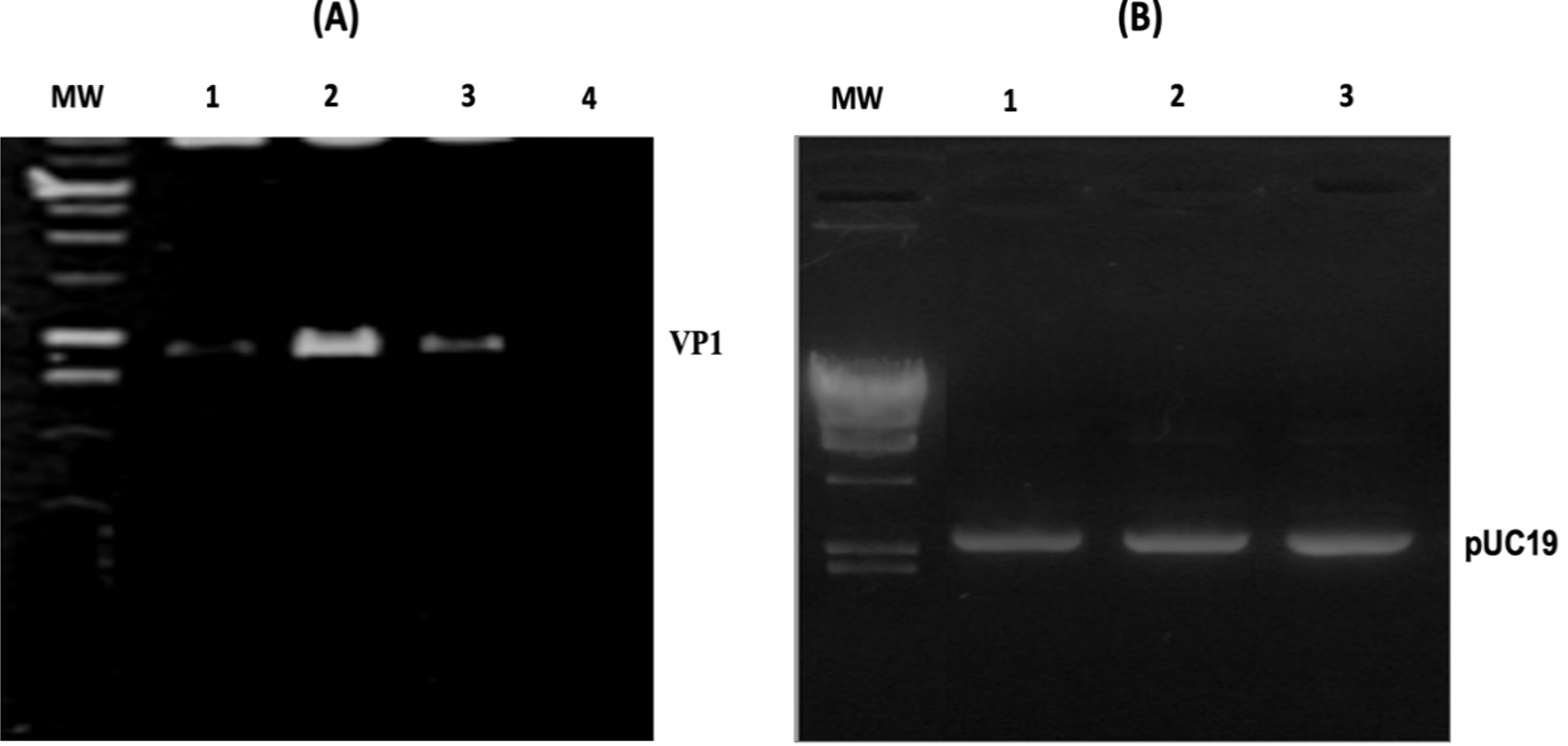
Figure 4. Revelation on 1% agarose gel of PCR colony products and plasmids extraction. (A) The MW track corresponds to the size marker (50-1000 bp), and the tracks from 1 to 3 each correspond to the amplification products of the colony PCR reaction carried out on three different colonies. Track 4 represents the negative control. (B) Lane MW corresponds to the molecular weight (Phage Lamda DNA digested by HindIII), and lanes 1 to 3 correspond to the plasmids extracts from 3 different colonies
Sequencing of the VP1 cloned into pUC19 vector
For controlling the right and the good-orientated cloned gene, we performed a sequencing reaction of the recombinant plasmid by using primers targeting the plasmid backbone and the insert sequence. The products obtained were sequenced using an ABI PRISMTM 310 genetic analyzer sequencer (Applied Biosystems, MD, USA). Obtained DNA and corresponding Protein sequences of the rVP1 cloned in the plasmid pUC19 are presented in Table 1.
Table (1):
Comparison of the Physicochemical properties of rVP1 with the VP1 capsid protein of CVB3 Nancy
Parameters |
rVP1 |
VP1 |
|---|---|---|
Number of amino acids: |
284 |
287 |
Molecular weight: |
31709.55 |
32068.95 |
Theoretical pI: |
7.11 |
5.96 |
AA most present |
Thr (T): 9.5% Ser (S): 8.5% Ala (A): 7.4% |
Thr (T): 10.1% Ser (S): 8.4% Ala (A)& Val (V): 7.3% |
Atomic composition |
C1419H2146N382O427S10 |
C1434H2167N383O434S11 |
Total number of atoms: |
4384 |
4429 |
Estimated half-life 1: (Escherichia coli, in vivo). |
2 min |
2 min |
Extinction coefficients are measured in water at 280 nm and are expressed in M-1 cm-1 units. |
48610 48360 |
48735 48360 |
Instability index2 |
44.54 |
41.79 |
Aliphatic Index |
67.99 |
69.30 |
Grand average of hydropathicity (GRAVY):3 |
-0.292 |
-0.246 |
1Half-life conditions (rVP1 N-terminal was W (Trp), VP1 N-terminal F (Phe), VP2 N-terminal T (Thr), VP3 N-terminal Y (Tyr) and VP4 N-terminal G (Gly).
2An instability index <40 indicates stability and an index >40 indicates instability.
3The average hydrophilic coefficient ranges from –2 to 2, a positive value indicates low hydrophilicity and negative values indicate high hydrophilicity.
Molecular characterization of the produced rVP1 by SDS-PAGE
After producing more recombinant bacteria to obtain more recombinant proteins, we successfully extracted these proteins by the sonication method. Then, we performed the separation of proteins by SDS-PAGE. With this method, the proteins are entirely denatured and given a negative charge by sodium dodecyl sulfate (SDS). The applied negative charge in the electric field separates them according to size. After the gel showed a dark blue color, it was transferred to a distaining solution until the bands were visible. Bands for three different positive colonies with rVP1 were obtained on SDS-PAGE gel (Figure 5). The location of the bands was around 30 kDa.
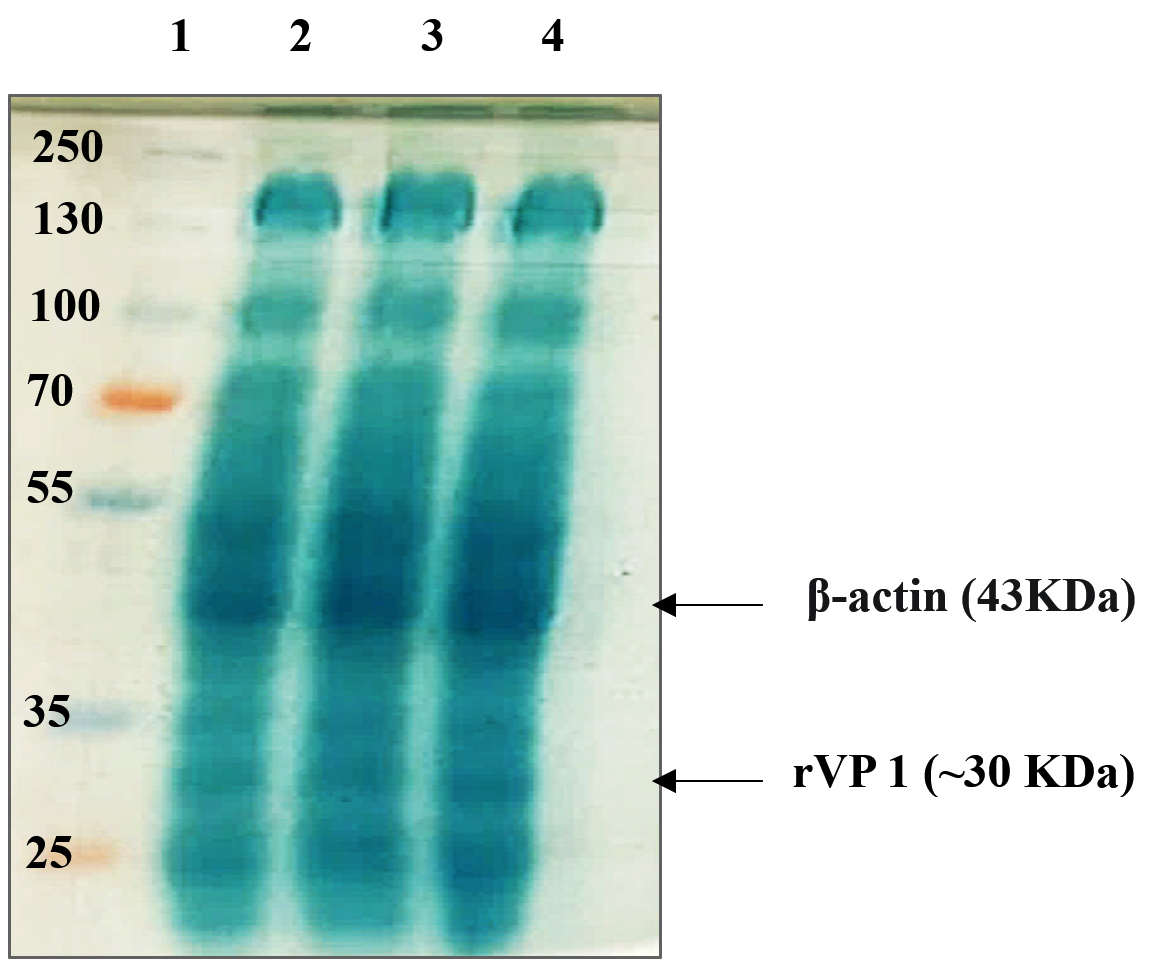
Figure 5. SDS-PAGE analysis of the rVP1 protein. Lane 1 corresponds to the molecular weight marker. Lanes 2 to 4 correspond to the rVP1 extracts from 3 different colonies. Arrow showed rVP1 bands, number on the left showed the marker size of protein molecular weight (kDa)
Bioinformatics analysis of the sequence and the structure of the viral rVP1
Genomic analysis of the nucleotide sequence of rVP1
To be sure of the nucleotide sequence obtained, we compared the DNA sequence of our strain with the VP1 of the CVB3 wild type Nancy strain. The Clustal Omega tool (https://www.ebi.ac.uk/Tools/msa/clustalo/) was used to compare the sequences, and the results showed that the VP1 gene cloned in plasmid pUC19 is 97.11% identical to the CVB3 Nancy strain (Figure 6). According to the analysis by the “cdd” database (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi), our insert will be expressed like all the structural proteins in a protein of the capsid of the viral family “Picornaviridae”.

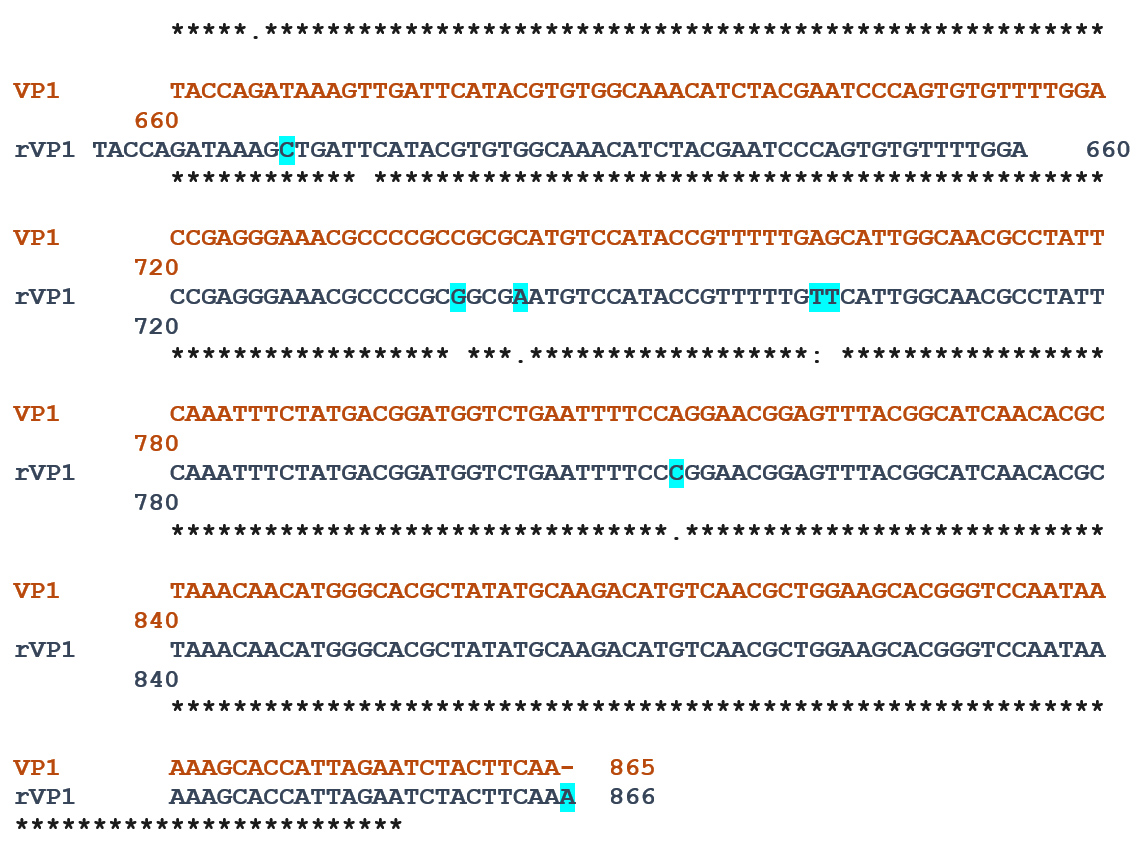
Figure 6. Sequence alignment between rVP1 and VP1 showed 97.11% of identity. The nucleotide differences between both sequences were highlighted in the blue background
Proteomic analysis of the rVP1
Using Sequence Manipulation Suite (SMS) (https://www.bioinformatics.org/sms2/translate.html), we translated the DNA sequences of the generated rVP1 into protein sequences. Next, we examined the physicochemical characteristics of the VP1 and rVP1 proteins of the CVB3 Nancy strain using the bioinformatics tool ProtParam of the ExPASy website (https://web.expasy.org/protparam/). These attributes are summarized in Table 1.
We used bioinformatics tools to find the phosphorylation sites in the recombinant nucleoprotein of the Coxsackievirus B3 virus. Phosphorylation of proteins or small molecules is a post-translational event that occurs in the cytosol or cell nucleus. This chemical modification consists of adding a phosphate group on a molecule taken from an ATP molecule which then becomes ADP on particular amino acids: serine, threonine, or tyrosine residues. This key event in the regulation of cellular processes such as metabolism, proliferation, differentiation, and apoptosis are carried out by members of the protein kinase family, the second family of the human genome.23 The phosphorylation sites in the recombinant nucleoprotein rVP1 were evaluated using a standard bioinformatics tool named “NetPhos Server” (https://services.healthtech.dtu.dk/services/NetPhos-3.1/). From this study, 37 phosphorylation sites were identified. Classifying by specific amino acid group, there were 13 specific serine sites, 19 specific threonine sites, and 5 tyrosine specific sites, with different phosphorylation potentials. Details for the positioning in the sequence and the potential for phosphorylation are shown in Figure 7.
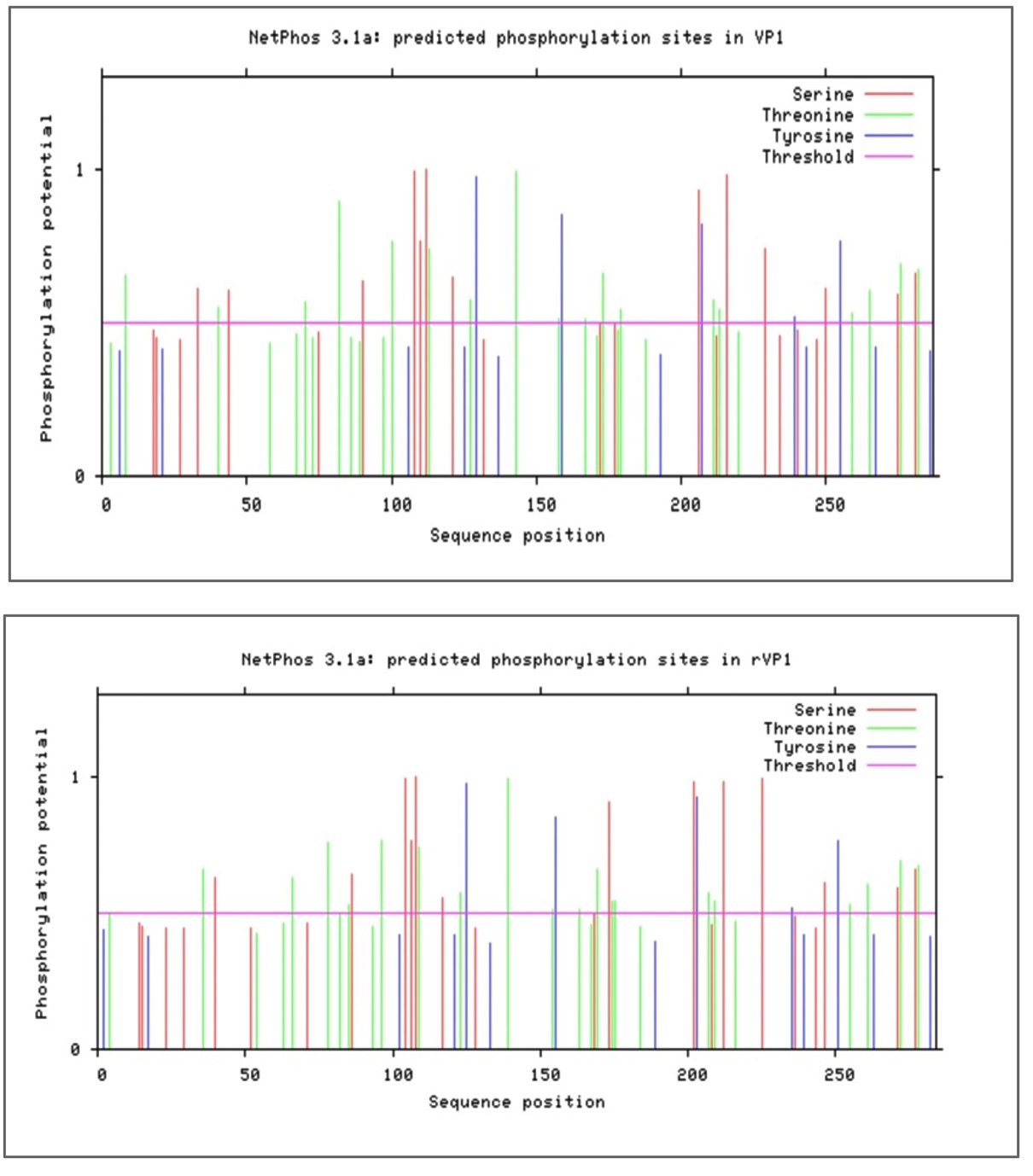
Figure 7. Prediction of the positions of the phosphorylation sites in VP1 and rVP1
Prediction and characterization of the secondary and tertiary structures of the rVP1
We predicted and compared the secondary structure of rVP1 and the VP1 of the CVB3 Nancy strain using SOPMA program (https://npsa-prabi.ibcp.fr/cgi-bin/npsa_automat.pl?page=/NPSA/npsa_sopma.html). The results are presented in Figure 8.

Figure 8. Prediction of secondary structures of the viral rVP1
SWISS-MODEL is a structural homology modeling server for proteins. This program (https://swissmodel.expasy.org/interactive) was used to predict homologous sequences. Proteins with high similarity and coverage with the sequence ratio are selected as templates for modeling. Table 2 indicates the model’s most homologous to each protein sequence rVP1 and VP1.
Table (2):
Prediction of homologous sequences to rVP1 and VP1 of CVB3 Nancy strain
Protein |
Template |
GMQE |
QMEAN |
|---|---|---|---|
rVP1 |
7vxh.1.A |
0.70 |
0.74 |
VP1 |
7vxh.1.A |
0.70 |
0.77 |
By using Cn3D Program (https://www.ncbi.nlm.nih.gov/Structure/icn3d/) we view the macromolecular structure that allows us to view 3-dimensional structures. The file type used in this study is the Protein Data Bank (PDB). Figure 9 presents the tertiary structures of the rVP1 and VP1 proteins.
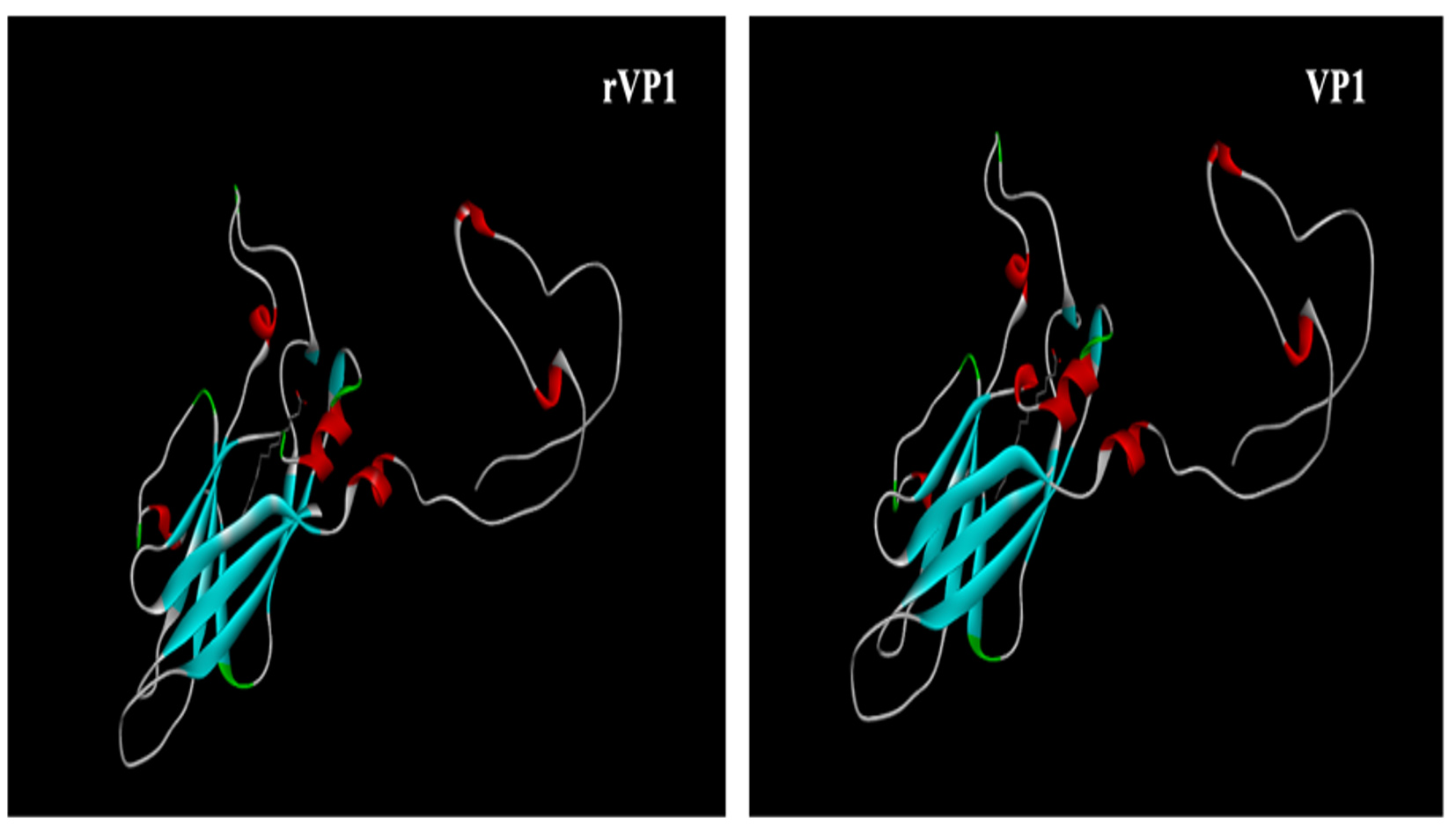
Figure 9. Prediction of the CVB3 Nancy strain’s rVP1 and VP1 tertiary structures. The ± helices are represented by the red stripes, the ² sheets by the light blue ones, and the other residues by the white ones
Prediction of the rVP1 epitopes
The Bepipred program (https://services.healthtech.dtu.dk/services/BepiPred-2.0/) is utilized to forecast the rVP1 epitopes, which are identified by the region recognized by the humoral reaction products, or antibodies. Significant peptides are composed of more than ten amino acids. VP1 and rVP1 have the highest potential B-type antigenic determinant sites and are highly similar (Table 3).
Table (3):
Potential sites for B epitopes in rVP1 and VP1 of the CVB3 Nancy strain
| Protein | No. | Start | End | Peptide | Length |
|---|---|---|---|---|---|
| VP1 | 1 | 39 | 59 | DTPFISQQNFFQGPVEDAITA | 21 |
| 2 | 62 | 98 | GRVADTVGTGPTNSEAIPALTAAETGHTSQVVPGDTM | 37 | |
| 3 | 102 | 111 | HVKNYHSRSE | 10 | |
| 4 | 130 | 138 | ENSGAKRYA | 9 | |
| 5 | 174 | 184 | QQPSTTQNQDA | 11 | |
| 6 | 199 | 212 | PVPDKVDSYVWQTS | 14 | |
| 7 | 223 | 224 | NA | 2 | |
| 8 | 246 | 258 | WSEFSRNGVYGIN | 13 | |
| 9 | 276 | 276 | T | 1 | |
| rVP1 | 1 | 35 | 52 | DTPFISQQNFFQGPVEDS | 18 |
| 2 | 61 | 94 | ADTVGTGPINSEAIPALTAAKTGHTSQVVPGDTM | 34 | |
| 3 | 98 | 108 | HVKNYHSRSES | 11 | |
| 4 | 127 | 133 | NSGAKRY | 7 | |
| 5 | 170 | 180 | QQPSTTQNKDA | 11 | |
| 6 | 195 | 208 | PVPDKADSYVWQTS | 14 | |
| 7 | 219 | 220 | NA | 2 | |
| 8 | 242 | 254 | WSEFSRNGVYGIN | 13 | |
| 9 | 271 | 273 | STG | 3 |
The analysis of the possible epitopes by Bepipred showed that VP1 contains 5 epitopes composed of more than 10 residues in aa, respectively located at aa 5 to 31, aa 39 to 59, aa 62 to 98, 174 to 184, 199 to 212 and aa 246 to 258. rVP1 contains 6 of them more or less close to those of VP1 which are located at level 35 at 52, 61 at 94, 98 at 108, 170 at 180, 195 at 208 and 271 at 273. All potential epitopes are exposed at the surface of viral proteins as shown in Figure 10.
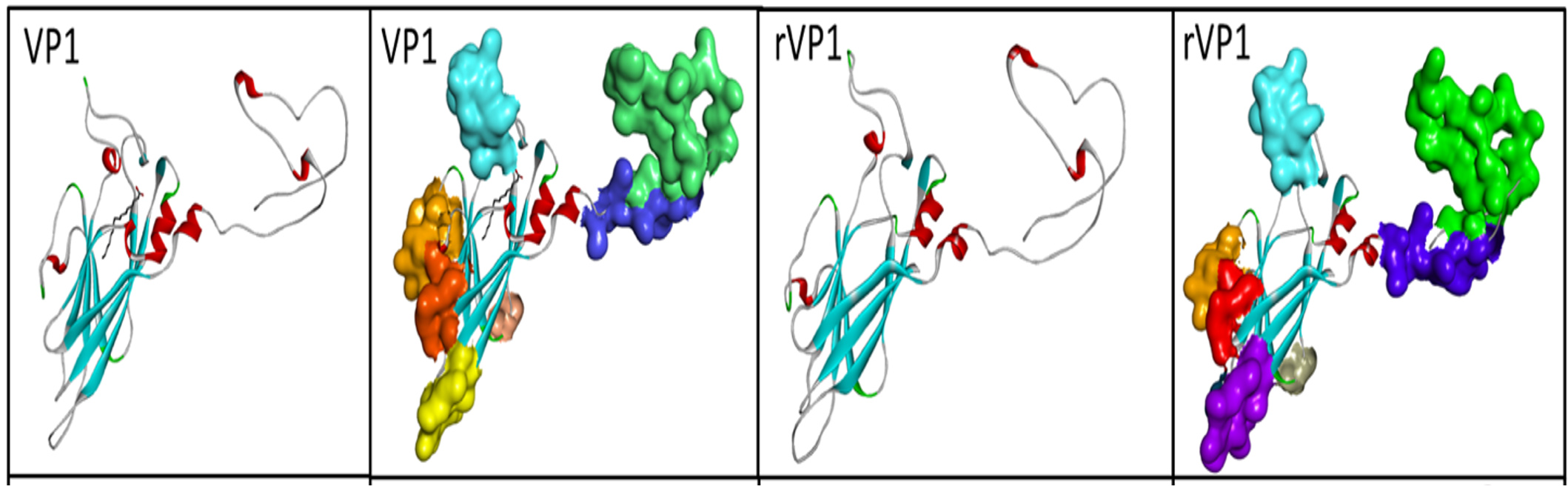
Figure 10. Possible B-epitope regions of proteins shown in surface view. The color spectrum for B epitopes includes red, blue, green, gold, purple, orange, and a light shade of green
CVB3 is a member of the Picornaviridae family and Enterovirus genus. It is a single-stranded RNA with positive strands that is shielded by a viral capsid. Its 7.4 kb RNA is segmented into P1, P2, and P3 sections. P1 is the first region and encodes VP1 through VP4, the four structural proteins. Most epitopes binding neutralizing antibodies are found on VP1, the largest and most changeable of these structural proteins. In addition, The VP1 proteins maintain the viral morphology of the virus but also contribute to CVB3 pathogenicity. Modifications to several VP1 amino acids reduce CVB3 virulence.24 These properties make VP1 an effective means for viral identification, infection control, and evolutionary analysis.25-27.
Based on this information, we have set the aim of cloning the VP1 gene in the pUC19 plasmid vector to gather as much data as possible for the experiments necessary for the development of vaccine and/or immunodiagnostic reagents. Once the target nucleotide sequence has been cloned and the outcome verified by insert sequencing, we applied the bioinformatics analysis to offer helpful data for future experimental studies and preparatory or preliminary research work on the produced recombinant VP1.
According to genomic study, there is no discernible sequence change between the VP1 recombinant gene and the CVB3 wild type Nancy strain, with the two sequences being 97% similar. This clearly shows that the cloned gene can be used to carry out any studies concerning the viral strains of CVB3. Then, the proteomic analysis revealed that the produced rVP1 protein has many identical or similar physicochemical properties and structural and functional domains to the wild type of strain VP1 but also different characteristics. Also, the two proteins with the most phosphorylation sites rVP1 and VP1 have 37 and 36 sites, respectively. Therefore, these proteins, especially VP1 and the produced recombinant protein rVP1 can affect host cell signal transduction. The structural characteristics of protein sequences, such as hydrophilicity, solvent accessibility, and flexibility, form the basis of antigenic determinants of proteins known as epitopes. Many epitope prediction protocols based on these features have been widely applied in vaccine development.28
Our study revealed that the VP1 and the rVP1 proteins were all hydrophilic; their secondary structure mainly contained random coils and had a relatively higher ratio of exposed aa residues. These results suggest that VP1 proteins possess all the attributes of antigenic epitopes.28 Features of the tertiary structure also supported the epitope prediction results. The Bepipred database predicted that rVP1 contains a more significant number of B epitopes than other proteins. Indeed, rVP1 has six possible antigenic determinants on the same nucleotide positions as VP1.
These results provide more insight into the pathogenicity of CVB3 via VP1, and they serve as a basis for subsequent research aimed at using this viral capsid protein to generate vaccines and/or associated immunodiagnostic reagents. The primary emphasis of the research effort presented in this paper is the CVB3 serotype, which plays a role in the pathophysiology of dilated cardiomyopathy, chronic myocarditis, and autoimmune illnesses including type 1 diabetes (T1D). To date, no vaccine or clinical treatment has been prescribed against this virus. To thwart the spread of the infection in the population, particularly newborns and the immunocompromised, vaccination against CVB3 is recommended. Based on this, we concluded that a vaccine against the CVB3 strain could eliminate infections associated with these diseases in humans.
Using the pUC19 expression vector, we were able to successfully produce the viral capsid protein VP1 gene of the CVB3 Nancy strain in E. coli DH5. A complementary bioinformatics investigation was employed to characterize the generated rVP1. We gathered crucial data for upcoming experimental investigations, preliminary or preparatory work for VP1 protein research, and the creation of vaccines and/or immunodiagnostic reagents. In fact, the genomic study revealed a significant homology between the VP1 recombinant gene and the CVB3-Nancy strain. The VP1 and rVP1 proteins have several structural and functional domains, as well as physicochemical characteristics, that are same or similar, according to proteomic study of the expressed rVP1. Furthermore, they possess a considerably larger ratio of exposed amino acid residues, are hydrophilic, and can influence the transmission of signals by host cells. These results provide more insight into the pathogenicity of CVB3 via VP1, and they serve as a basis for subsequent research aimed at creating a potential vaccine and/or associated immunodiagnostic techniques utilizing this recombinant viral capsid protein.
ACKNOWLEDGMENTS
The authors would like to thank the Deanship of Scientific Research, Vice Presidency for Graduate Studies and Scientific Research, King Faisal University, Saudi Arabia.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
AUTHORS’ CONTRIBUTION
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
FUNDING
This work was supported by the Deanship of Scientific Research, Vice Presidency for Graduate Studies and Scientific Research, King Faisal University, Saudi Arabia, with grant number 2610.
DATA AVAILABILITY
All datasets generated or analyzed during this study are included in the manuscript.
ETHICS STATEMENT
Not applicable.
- Geisler A, Hazini A, Heimann L, Kurreck J, Fechner H. Coxsackievirus B3-Its Potential as an Oncolytic Virus. Viruses. 2021;13(5):718.
Crossref - Chiu W-Y, Lo Y-H, Yeh T-C. Coxsackievirus associated hand, foot and mouth disease in an adult. QJM. 2016;109(12):823-824.
Crossref - Laga AC, Shroba SM, Hanna J. Atypical hand, foot and mouth disease in adults associated with coxsackievirus A6: a clinico-pathologic study. J Cutan Pathol. 2016;43(11):940-945.
Crossref - Lee KY. Enterovirus 71 infection and neurological complications. Korean J Pediatr. 2016;59(10):395-401.
Crossref - Fowlkes AL, Honarmand S, Glaser C, et al. Enterovirus Associated Encephalitis in the California Encephalitis Project, 1998-2005. J Infect Dis. 2008;198(11):1685-1691.
Crossref - Zhang L, Yan J, Ojcius DM, et al. Novel and Predominant Pathogen Responsible for the Enterovirus-Associated Encephalitis in Eastern China. PLoS ONE. 2013;8(12):e85023.
Crossref - Garmaroudi FS, Marchant D, Hendry R, et al. Coxsackievirus B3 replication and pathogenesis. Future Microbiol. 2015;10(4):629-653.
Crossref - Pettersen EF, Goddard TD, Huang CC, et al. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021;30(1):70-82.
Crossref - Honkimaa A, Kimura B, Sioofy-Khojine AB, et al. Genetic adaptation of coxsackievirus b1 during persistent infection in pancreatic cells. Microorganisms. 2020;8(11):1790.
Crossref - Linden LV, Wolthers KC, van Kuppeveld FJM. Replication and Inhibitors of Enteroviruses and Parechoviruses. Viruses. 2015;7(8):4529-4562.
Crossref - Andreioletti L, Renois F, Jacques J, Leiveque N. Enteirovirus non poliomyeilitiques et pathologies respiratoires. Med Sci. 2009;25(11):921-930.
Crossref - Huang B, Harrower B, Burtonclay P, Constantino T, Warrilow D. Genome Sequences of Coxsackievirus B5 Isolates from Two Children with Meningitis in Australia. Genome Announcements. 2017;5(41):e01125-17.
Crossref - Tuthill TJ, Groppelli E, Hogle JM, Rowlands DJ. Picornaviruses. Curr Top Microbiol Immunol. 2010;343:43-89.
Crossref - Hassine IH, Gharbi J, Hamrita B, Almalki MA, Rodriguez JF, ben M’hadheb M. Characterization of Coxsackievirus B4 virus-like particles VLP produced by the recombinant baculovirus-insect cell system expressing the major capsid protein. Mol Biol Rep. 2020;47(4):2835-2843.
Crossref - Xiang P, Mohamud Y, Luo H. SNAP47 Interacts with ATG14 to Promote VP1 Conjugation and CVB3 Propagation. Cells. 2021;10(8):2141.
Crossref - Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162(1):156-159.
Crossref - Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A. 1977;74(12):5463-5467.
Crossref - Chung CT, Niemela SL, Miller RH. One-step preparation of competent Escherichia coli: transformation and storage of bacterial cells in the same solution. Proc Natl Acad Sci U S A. 1989;86(7):2172-2175.
Crossref - Blom N, Gammeltoft S, Brunak S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J Mol Biol. 1999;294(5):1351-1362.
Crossref - Blom N, Sicheritz Ponten T, Gupta R, Gammeltoft S, Brunak S. Prediction of post translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics. 2004;4(6):1633-1649.
Crossref - Letunic I, Bork P. 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 2018;46(D1):D493-D496.
Crossref - Letunic I, Doerks T, Bork P. SMART: recent updates, new developments and status in 2015. Nucleic Acids Res. 2015;43(D1):D257-D260.
Crossref - Keck F, Ataey P, Amaya M, Bailey C, Narayanan A. Phosphorylation of Single Stranded RNA Virus Proteins and Potential for Novel Therapeutic Strategies. Viruses. 2015;7(10):5257-5273.
Crossref - Zhou Y, Zhang Z, Wang H, et al. In vitro interaction between coxsackievirus B3 VP1 protein and human pleckstrin homology domain retinal protein (PHR1). Virus Genes. 2015;51(2):182-189.
Crossref - Henke A, Wagner E, Whitton JL, Zell R, Stelzner A. Protection of Mice against Lethal Coxsackievirus B3 Infection by Using DNA Immunization. J Virol. 1998;72(10):8327-8331.
Crossref - Henke A, Jarasch N, Wutzler P. Coxsackievirus B3 vaccines: use as an expression vector for prevention of myocarditis. Expert Rev Vaccines.2008;7(10):1557-1567.
Crossref - Yuan S, Li G, Wang Y, et al. Identification of Positively Charged Residues in Enterovirus 71 Capsid Protein VP1 Essential for Production of Infectious Particles. J Virol. 2016;90(2):741-752.
Crossref - Soria-Guerra RE, Nieto-Gomez R, Govea-Alonso DO, Rosales-Mendoza S. An overview of bioinformatics tools for epitope prediction: Implications on vaccine development. J Biomed Inform. 2015;53:405-414.
Crossref
© The Author(s) 2024. Open Access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License which permits unrestricted use, sharing, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.