


ISSN: 0973-7510
E-ISSN: 2581-690X
In modern drug discovery, molecular docking analysis is routinely used to understand and predict the interaction between a drug molecule and a target protein from a microbe. Drugs identified in this way may inhibit the entry and replication of pathogens in host cells. The SARS-CoV-2 associated coronavirus disease, COVID-19, has become the most contagious and deadly pandemic disease in the world today. In abeyance of any specific vaccine or therapeutic against SARS-CoV-2, the burgeoning situation urges a need for effective drugs to treat the virus-infected patients. Herbal medicines have been used as natural remedies for treating various infectious diseases since ancient times. The spike (S) protein of SARS-CoV-2 is important for the attachment and pathogenesis of the virus. Therefore, this study focused on the search of useful ligands for S protein among active constituents present in common herbs that could serve as efficient remedies for COVID-19. We analysed the binding efficiency of twelve compounds present in common herbs with the S protein of SARS-CoV-2 through molecular docking analysis and also results are validated with two different docking tools. The binding efficiency of ligands was scored based on their predicted pharmacological interactions coupled with binding energy estimates. In docking analysis, compound “I” (Epigallocatechin gallate (EGCG)) was found to have the highest binding affinity with the viral S protein, followed by compounds, “F” (Curcumin),“D” (Apigenin) and “E” (Chrysophanol). The present study corroborates that compound “I” (EGCG) mostly present in the integrants of green tea, shows the highest potentiality for acting as an inhibitor of SARS-CoV-2. Further, characterization of the amino acid residues comprising the viral binding site and the nature of the hydrogen bonding involved in the ligand-receptor interaction revealed significant findings with herbal compound “I” (EGCG) binding to the S protein at eight amino acid residues. The binding sites are situated near to the amino acids which are required for virus pathogenicity. The findings of the present study need in vivo experiments to prove the utility of “I”, “F”,“D” and “E” compounds and their further use in making herb-based anti-SARS-CoV-2 product in near future. This analysis may help to create a new ethno-drug formulation for preventing or curing the COVID-19.
SARS-CoV-2, COVID-19, spike protein, molecular docking, in-silico, ligands, herbal medicine
The World Health Organization (WHO) defines traditional medicine as: “the sum of total knowledge, practices, and skills based on the historical theories, beliefs, and experiences…in indigenous to various cultures that are used to maintain the human or animal health and to prevent, diagnose, improve, or treat physical/mental illnesses” 1.
Herbal remedies are widely used in both developed and developing world countries to treat various illnesses indispensable 2. The WHO reported, about 80% of the world’s population depends primarily on traditional medicine to treat their illnesses. Traditional medicine is often considered to be a kind of complementary or alternative medicine (CAM)3.Herbal medicines include herbs, herbal preparations, and finished herbal products (tea varieties), as well as additives derived from different kinds of herb/plant parts (ginger, garlic, lemon, and so on), which are used when preparing food in many Asian countries, including India and China. The active components of these herbs have many advantages, like lower toxicity and allergenicity than some commercial medications, regulating immunological responses, and causing viral destruction4. Various common herbs have been used to prevent viral infections, and their efficacy has been demonstrated in research trials5,6. Herbal plants like Bupleurum spp., Heteromorpha spp., and Scrophularia scorodonia have been used in the treatment of coronaviruses in China7, and Azadirachta indica, Carica papaya, and Hippophae rhamnoides have been scientifically proven to be effective in treating or preventing Dengue fever in India8. Therefore, identifying and documenting the herbs that are effective in treating contagious diseases is vital for future disease control programs.
Since December 2019, novel coronavirus disease (COVID-19) has been spreading globally from its initial epicenter Wuhan, China. It causes severe respiratory problems more often in children and older people, who have weaker immune defences. The agent, responsible for the infection, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), belongs to the Betacoronavirus genus in the family Coronaviridae, the members of which infect a wide range of hosts9,10. Most of the people affected with SARS-CoV-2 experience moderate respiratory illness, from non-pneumonia to mild pneumonia in nature, along with headache, runny nose, and fever11. However, older and comorbid people who suffer from cardiovascular diseases, diabetes, and chronic respiratory diseases are more likely to develop severe symptoms (dyspnea, respiratory failure, septic shock, and multiple organ dysfunction/failure). Recently, some other symptoms like bluish spots on the feet, clotting, and stroke also noticed in COVID-19 positive patients12.
SARS-CoV-2 is a single-stranded, positive-sense RNA virus; its genome is 29.891 kb in size, enclosed by a 5′-cap and 3′poly-A tail, and has a G + C content of 38%. The virion is encircled with an envelope containing viral nucleocapsid and arranged with helical symmetry13. The SARS-CoV-2 genome encodes four major structural proteins, specifically the spike (S), membrane (M), envelope (E), and nucleocapsid (N) proteins. Among the proteins of similar coronaviruses, the S proteins of Severe acute respiratory syndrome-related coronavirus (SARS-CoV) and Middle East respiratory syndrome-related coronavirus (MERS-CoV) play an important role in binding with host cellular receptor, angiotensin-converting enzyme 2 (ACE2) and subsequent membrane fusion. Unlike non-structural proteins, the S protein is the major antigen responsible for inducing effective neutralizing antibodies to block viruses from binding to the host cells, and thus, inhibit viral infection14,15.
Molecular docking studies are primarily carried out to estimate how two or more molecules interact with each other and can depict the best-fit orientation of a ligand (a small drug-like molecule) that binds to a target protein. The most significant use of this approach is in determining the protein-ligand interaction due to its applications in drug discovery. Molecular docking analysis is a bioinformatics-based approach to analyse the fit, binding, and interactions between protein and ligand, based on their binding energy. The results of such interaction studies are presumed to allow the prediction of suitable ligands for drug production, which can then be used in a particular pharmaceutical treatment approach16,17. The current study aimed to compare the docking fit of various selected compounds from indigenous food additives and herbal constituents with the SARS-CoV-2 S protein and find the best-fitting components. Also, we characterized the amino acid residues comprising the viral binding site and the nature of the hydrogen bonding involved in the ligand-receptor interaction. This analysis may help to create a new ethno-drug formulation for preventing or curing COVID-19.
Selection of viral target protein
Because of its importance in viral pathogenesis, S protein was targeted in this study. We attempted to predict the major ligands for the viral S protein using indigenous food additives and herbal constituents. Information on the 3D structure of the viral S protein (PBD ID: 6VYB) was obtained from the protein data bank (PDB) and used in the analyses. Protein structures and docking were visualized using RasMol 2.7.3 software18.
Selection and preparation of ligands
Information on the different herbal ligands selected in this study was based on the review of previous researches, and the details of the compounds in them and the herbs from which they were retrieved are given in Table 1. A total of 12 ligands were selected for testing. Information on the 3D structures of all 12 ligands were downloaded from PubChem19in structure-data file (SDF) format and converted into MDL Molfile (MOL) format with the help of Open Babel Server20, and data in this format was used as input for Generic Evolutionary Method for Molecular Docking (iGEMDOCKv2.1) analyses21.
Table (1):
Details of the tested herbal components, along with their active ingredients and PubChem CIDs. The table also depicts, the known function of each compound, the native of origin, and associated references from PubMed-NCBI.
Code |
Herb |
Active ingredient |
PubChem CID |
Function of Compounds |
Origins |
References |
|---|---|---|---|---|---|---|
A |
Allium sativum |
Ajoene |
5386591 |
Anticancer activity |
Central Asia and northeastern Iran |
27 |
B |
Allium sativum |
Allicin |
65036 |
Antibacterial and anti-fungal properties |
Central Asia and northeastern Iran |
28 |
C |
Aloe barbadensis |
Aloe emodin |
10207 |
Interferon (IFN)-inducer, antiviral activity of aloe-emodin against Japanese encephalitis virus, enterovirus and influenza virus |
South-west Arabian Peninsula |
29,30 |
D |
Petroselinum crispum |
Apigenin |
5280443 |
Anti-viral agents, anti-tumor, bowel disease and skin conditions |
central and eastern Mediterranean region |
31 |
E |
Aloe barbadensis |
Chrysophanol |
10208 |
Antiviral and anti-inflammatory activity |
South-west Arabian Peninsula |
32 |
F |
Curcuma longa |
Curcumin |
969516 |
Antioxidant and anti-inflammatory effects |
Southwest India |
33 |
G |
Allium sativum |
Diallyltrisulfide |
16590 |
Antineoplastic agent, an antifungal agent |
Central Asia and northeastern Iran |
34 |
H |
Aloe barbadensis |
Emodin |
3220 |
Antimicrobial and laxative activity |
South-west Arabian Peninsula |
35 |
I |
Camellia sinensis |
Epigallocatechin gallate |
9804842 |
Antioxidant and Antimicrobial Activities |
Southeast Asia |
36 |
J |
Zingiber
officinale |
Gingerol |
5317599 |
Antioxidant, anti-tumor and anti-inflammatory |
Tropical Asia |
37 |
K |
Malus domestica |
Ursolic acid |
64945 |
Antiparasitic activity, antitumor, and immunomodulatory property. |
Temperate areas of the world |
38;39 |
L |
Zingiber officinale |
Zingerone |
31211 |
Appetite stimulant, anxiolytic, antithrombotic, radiation protective, and antimicrobial. |
Tropical Asia |
40 |
Molecular docking analyses with iGEMDOCK
The graphical-automatic drug design system iGEMDOCK was used for in silico docking, screening, and post-docking analyses. The S protein structure file was uploaded to the Prepare Binding Site server, and the “By Current File” option was selected so that the overall uncut protein surface could be checked for binding. The ligands were loaded using the “Prepare Compounds” option, and a library of ligands was then prepared for the analyses.
Default docking parameters were used for testing the docking performance of the ligands with the S protein of SARS-CoV-2 (population size=200, number of generations =70, and number of solutions =2). Standard docking was performed with the iGEMDOCK scoring function with ligand and electrostatic preferences set at 1.00. To get accurate and speed up the process, standard docking was used as a default setting. To avoid false positive or false negative results, four rounds of docking was carried out with the same protein and ligands as reported earlier41,42. In these analyses, a lower energy profile indicated a more stable interaction, and the lowest energy profile represented the most likely binding interaction between the protein and ligand tested. After the docking process, the best docking position for each of the individual ligands relative to the S protein was analysed. The outputs obtained from the docking analyses, including the binding position, binding energy, van der Waals force, and hydrogen bonding energy values, were then retrieved, and the best-fitting 3D conformation was analysed with RasMol Viewer18,21.
Molecular docking analysis with AutoDock software
This software composed of two different tools such as AutoDock Tool 1.5.6 and AutoDockVina 1.1.2 and works based on the Lamarckian genetic algorithm. Initially, AutoDockTool was used to optimize and prepare the target protein and ligands in a format of pdbqt files. The Actual docking process was carried out with AutoDockVina software, in which binding site of protein was adjusted with the use of grid box and x, y, z axis values changed to 220 X 210 X 180. The grid point spacing was adjusted to 0.400 Å. The conformational similarity of docked poses was estimated by calculating the root mean square deviations (RMSD) values. RMSD values and the lowest binding energy of conformations is the most stable docking pose43. The output files from AutoDockVina were analysed with PyMOL 2.3.4 trial version software44. Finally, the molecular docking results of the iGEMDOCK software were compared with AutoDockVina software outputs.
In this study, the 3D structure model of the SARS-CoV-2 S protein was optimized and 12 ligands from previous studies and online resources were selected to test them as binding ligands for the S protein. The 3D structures of these ligands were also retrieved and optimized for docking analysis. The total binding energy for all the herbal ligands was calculated using iGEMDOCK software, and the binding conformations of the tested ligands with the S protein were also evaluated. From the docking analyses, the binding affinities of twelve compounds to the S protein were estimated based on their estimated ligand binding energy; the results are listed in Table 2. The binding position for each ligand molecule relative to the S protein was analysed, and the one with the lowest ligand binding energy with the S protein, among the various positions tested (four rounds of docking runs), was identified as the most probable binding position. The lowest energy score indicated the ligand for which the protein-ligand binding affinity was the highest, while higher energy values indicated lower binding affinity. Among the 12 herbal ligands, four compounds, “I”,“F”, “D”, and “E” were found to have lowest binding energy values than the other ligands, and thus, had the highest binding affinity (Highlighted in Table 2). Compound “I” (EGCG) had the lowest binding energy value with the S protein (-130.566 kcal/mol), followed by compounds “F” (Curcumin), “D” (Apigenin) and “E”(Chrysophanol) with binding energy values -115.198 kcal/mol, -108.614 kcal/mol and-107.385 kcal/mol, respectively. The other compounds tested, such as “H”, “L”, “J” and “K” had a moderate binding affinity with the S protein ranging from -105.462 kcal/mol for Emodin (H) to -89.9499 kcal/mol for Urosilic acid (K).
Table (2):
The fit and interaction profiles of the tested ligands with the viral spike protein.The total binding energy, van der Waal’s force, H-bond energy, electrostatic force, and AverConPair of the binding of each ligand with the S protein are shown. The values shown below for each compound correspond to the lowest binding energy obtained among the four rounds of docking runs.
Code |
Compound name |
Total binding energy (kcal/mol) |
Van der Waal’s force (kcal/ mol) |
H-bond energy (kcal/mol) |
Electro static force (kcal/mol) |
AverCon Pair (kcal/mol) |
|---|---|---|---|---|---|---|
A |
Ajoene |
-74.2819 |
-68.2819 |
-6 |
0 |
32.4615 |
B |
Allicin |
-62.4326 |
-62.4326 |
0 |
0 |
40.3333 |
C |
Aloe emodin |
-69.2503 |
-69.2503 |
0 |
0 |
29.2308 |
D |
Apigenin |
-108.614 |
-82.1108 |
-26.503 |
0 |
33.9 |
E |
Chrysophanol |
-107.385 |
-90.5916 |
-16.7935 |
0 |
36.7895 |
F |
Curcumin |
-115.198 |
-87.5695 |
-27.6288 |
0 |
26.963 |
G |
Diallyltrisulfide |
-53.2872 |
-53.2872 |
0 |
0 |
39.2222 |
H |
Emodin |
-105.462 |
-87.2314 |
-18.2303 |
0 |
32.85 |
I |
Epigalloca techingallate |
-130.566 |
-91.7244 |
-38.8417 |
0 |
25.7273 |
J |
Gingerol |
-98.0333 |
-84.1818 |
-13.8515 |
0 |
27.6667 |
K |
Ursolic acid |
-89.9499 |
-72.2658 |
-17.6841 |
0 |
23.5714 |
L |
Zingerone |
-102.184 |
-77.9523 |
-24.2321 |
0 |
26.9524 |
Besides, the analyses also revealed the characteristics of binding H-bonds and the involved energy of the 12 herbal compounds with the target S protein. Docking position analyses showed the amino acids within the S protein involved in binding with each of the ligands “I”,“F”, “D”, and “E”. The Compound “I” is forming a hydrogen bond with spike protein at eight sites (GLN314, ASN317, ASP737, ASN764, THR859, THR315, VAL736 and ASP737). The 3D image of protein-ligand (“I”) interaction shown in Fig. 2. The energy of H-bonds typically ranges from -8.7 to -1.1 kcal/mol. The compounds “F, “D”, “E” formed hydrogen bonds at six, eight, and four sites, with H-bond energies from -7 to -1.1 kcal/mol, respectively. The details of the binding energy of each ligand, associated bonds, energy, and involved amino acid positions in S protein CDS are shown in Table 3 and Fig. 1.
Table (3):
H-bond and Amino acid position profile table for the spike protein with tested ligands. The total binding energy, H-bond nature, position of amino acids, and H-bond energy of each bond are shown.
| Code | Compound name | Total binding energy (kcal/mol) | H-bond | Amino acid positions | H-bond energy (kcal/mol) |
|---|---|---|---|---|---|
| A | Ajoene | -74.2819 | H-S | GLN1010,THR1009 | -3.5,-2.5 |
| H-M | |||||
| B | Allicin | -62.4326 | H-S | ||
| H-M | |||||
| C | Aloe emodin | -69.2503 | H-S | ||
| H-M | |||||
| D | Apigenin | -108.614 | H-S | LYS1038,ASP-1041 | -3.11991,-2.5, |
| H-M | GLY908,ASP-1041,GLY-1046,HIS-1048,TRP-886,ALA-890 | -2.5,-3.5, -3.56019,-1.12739,-2.5,-3.5, | |||
| E | Chrysophanol | -107.385 | H-S | ASP1041 | -2.5 |
| H-M | ASP1041,GLY1044,GLY1046 | -3.5,-2.5,-4.3 | |||
| F | Curcumin | -115.198 | H-S | ASN-544,ARG-567,ASN-978, ASP-979 | -3.5,-7,-3.5,-5.44359 |
| H-M | ALA-522,THR-547 | -3.5,-2.5, | |||
| G | Diallyltrisulfide | -53.2872 | H-S | ||
| H-M | |||||
| H | Emodin | -105.462 | H-S | LYS1038 | -3.5, |
| H-M | GLY908,HIS1048 | -2.5,-8.7 | |||
| I | Epigallocatechin gallate | -130.566 | H-S | GLN314,ASN317,ASP737, ASN764,THR859 | -6,-3.5,-3,-3.5,-2.5 |
| H-M | THR315,VAL736,ASP737 | -2.5,-5.3,-3.5 | |||
| J | Gingerol | -98.0333 | H-S | HIS-1058 | -5.35146 |
| H-M | ALA-1056,GLY-1059 | -6,-2.5 | |||
| K | Ursolic acid | -89.9499 | H-S | HIS1058, | -7.8 |
| H-M | LEU861,HIS1058 | -2.5,-6.7 | |||
| L | Zingerone | -102.184 | H-S | ASN978,ARG1000,THR573 | -3.4,-6.3,-2.5 |
| H-M | TYR741,GLY744,THR573 | -2.5,-3.2,-3.5 |
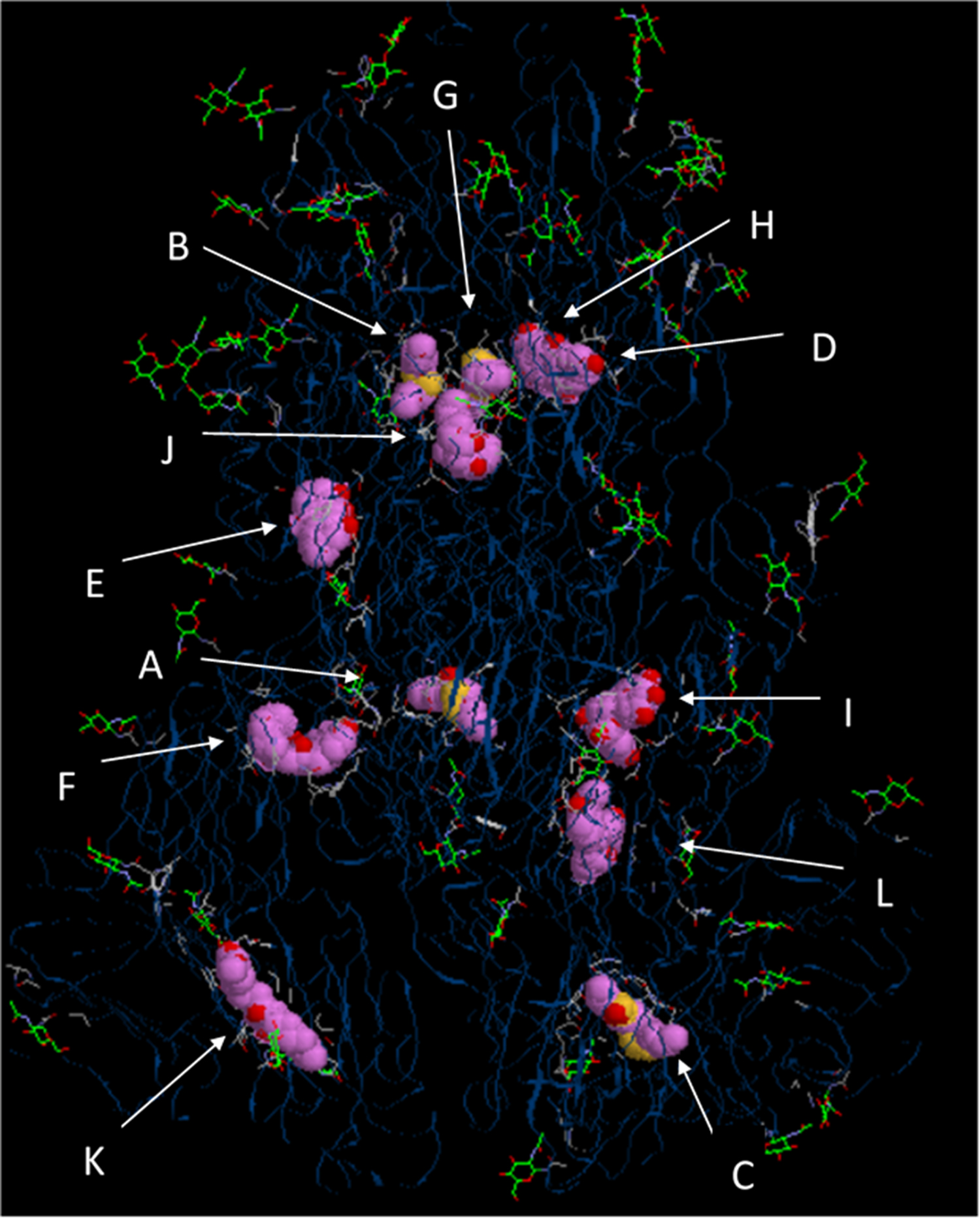
Fig. 1. Interactions of all tested herbal compounds with the spike protein of SARS-CoV-2, combined into a single figure. Interaction of various compounds to protein is as follows: A, Ajoene; B, Allicin; C, Aloe emodin; D, Apigenin; E, Chrysophanol; F, Curcumin; G, Diallyltrisulfide; H, Emodin;I, Epigallocatechingallate; J, Gingerol; K, Ursolic acid; L, Zingerone.
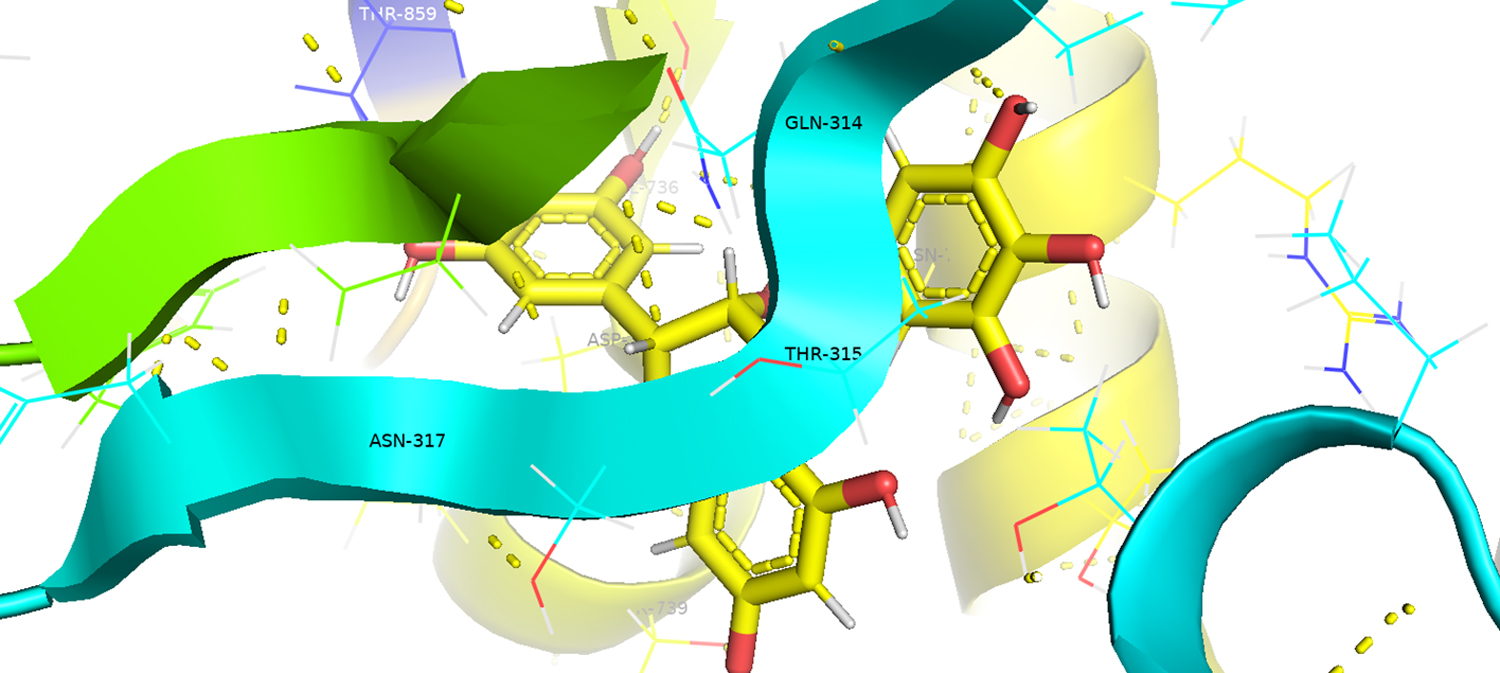
Fig. 2. PyMol software-based 3D image of spike protein –Epigallocatechingallate interaction showing H bond position of amino acids from spike protein and ligand are depicted. The amino acid residues of S protein involved in ligand binding (<500 amino acids positions) are highlited with black labelling.
AutoDockVina docking tool was used to compare and validate the results of iGEMDOCK output. The same spike protein and four compounds, which are having a high binding affinity with target protein based on iGEMDOCK were selected for comparison. Based on RMSD values, best poses were selected from the group of poses. The conformation which is having RMSD value lesser that 2.0Å is considered as the best pose. The RMSD value is used to measure the distance between two atoms in various protein-ligand conformations. Therefore best pose (<2.0Å) with the lowest binding energy poses are selected for further analyses. The result obtained from the AutoDockVina is mentioned in the Table No. 4. The AutoDockVina software results showed compound “I” having better affinity (-9.2 Kcal/mol) to the target protein. The other three compounds also showed a similar binding affinity like iGEMDOCK docking analysis.
Table (4):
Details of selected compound names and their respective affinity value obtained from the AutoDockVina given in the table.
Compound code |
Compound name |
Affinity (Kcal/mol) |
|---|---|---|
I |
Epigallocatechingallate (EGCG) |
-9.2 |
F |
Curcumin |
-8.2 |
D |
Apigenin |
-8.5 |
E |
Chrysophanol |
-8.6 |
COVID-19 has spread to nearly 210 countries with nearly 5 million confirmed cases and 325,000 deaths. Presently, the case fatality rate caused by the contagion seems to be lower in Asian region than European, American, or the world. Given this, we considered people’s food habits and identified a few common medicinal ingredients that are currently used in food preparation in India and some neighboring countries. Several drug candidates have been evaluated for their antiviral activity against SARS-CoV-2virus. But recent studies used chemical libraries to screen out better drug candidates for SARS-CoV-2 virus with limited success. So with this sense, we started to search herbals and herbal components to inhibit viral infections. Based on the literature, we selected twelve compounds that are used routinely in our lifestyle. Among 12 ligands, iGEMDOCK tool identified the best fitting ligand as EGCG (“I”) followed by Curcumin (“F”) Apigenin (“D”), and Chrysophanol (“E”) towards spike protein.
All the results based on iGEMDOCK were cross-verified and validated by comparing with another molecular modeling simulation software AutoDockVina which applies a different algorithm. Similar to iGEMDOCK tool, EGCG (“I”) showed highest binding affinity (-9.2 kcal/mol) as compared to other compounds. Thus the superiority of EGCG (“I”) was corroborated based on two docking approaches.
These compounds are having a better binding with spike protein in the form of H-bonding and van der Waal’s force. The compound EGCG is binding to spike protein with eight amino acids through H-bonding. The amino acids involved in the binding of proteins to the ligand are mostly polar. And the binding sites of ligands to S protein is essential for the virus binding to the ACE2 receptors. Babcock and co-workers22 reported that the amino acids positioned at the 270-510 are essential for the SARS-CoV-2 virus to attach with the host cell ACE2 receptor. Three amino acids from this region (GLN-314;THR-315;ASN-317) were found to be involved in ligand binding (Table 3; Fig. 2). Since the compounds, we tested block these amino acids, which may limit the invasion of host cells by SARS-CoV-2.
Binding energy estimates and binding site analyses of all ligands showed that binding positions of these amino acids mostly differ in their sites at which they formed hydrogen bonds. These hydrogen bonds are necessary to maintain the structural stability of the protein-ligand complex. Additionally, the H-bond energy of all compounds was nearly always negative, which indicated that they had a relatively high binding affinity with the S protein. Compound “I” which is mostly present in the components of green tea, has a high probability of blocking the virus attachment and entry into host cells. Similarly, previous studies have also shown that this compound can bind with the influenza virus23, Zika virus24, and porcine circovirus outer proteins22, thereby inhibiting viral entry into the host cells. This compound has the highest binding affinity towards SARS-CoV-2 spike protein compared to the antiviral drugs. Calligari and colleagues45 investigated the binding affinity of antiviral drugs with SARS-CoV-2 spike protein. In comparison to antiviral drug affinity, these herbal compounds showed better binding affinity.
Currently, many researchers are working on identifying candidate drugs via in silico analyses. The utilization of available antiviral medications might prove to be an effective method of inhibiting SARS-CoV-2 through the binding of these herbal ligands with the S glycoprotein, as well as the 3CL protease25. Rane and coworkers26 analysed the potential utility of various phytochemicals as ligands for the viral S protein, as determined by molecular docking study. The present study is perhaps the first to apply a molecular docking approach to predict both the potential ligand binding efficiency among the common herb/food constituents and to further elucidate the involved amino acids of SARS-CoV-2 S protein during ligand binding. This study divulged that among the tested ingredients, based on having the lowest estimated ligand binding energy, EGCG (the active ingredient in Camellia sinensis) was the most potent ligand against the S protein of SARS-CoV-2. Global scientific community mainly focused on developing antiviral drugs rather than finding any compounds which are upregulating the immune system46. This green tea compound EGCG also has many health benefits particularly modulating both adaptive and innate immune system functions47,48. Therefore, compounds that are used for treating illness, should have some additional immune regulatory functions which are worthwhile.
The molecular docking analyses helped to explore the probable binding modes of twelve ligands with the S protein of SARS-CoV-2. Among the tested compounds, EGCG (“I”), a principal constituent of green tea, had the highest binding affinity to this protein. Therefore, including green tea in the diets of people might help to reduce the occurrence of COVID-19. However, more in vivo experimental research is required to validate our results and for developing more potent drugs for the prevention and control of COVID-19.
ACKNOWLEDGMENTS
All the listed author(s) are thankful to their representative universities/institutes for providing the related support to compile this work.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
AUTHORS’ CONTRIBUTION
All the listed author(s) have made a substantial, direct, and intellectual contribution to the work, and approved it for publication.
FUNDING
None.
ETHICS STATEMENT
This article does not contain any studies with human participants or animals performed by any of the authors.
AVAILABILITY OF DATA
Not applicable.
- Qi Z. WHO traditional medicine strategy 2014-2023. Geneva: World Health Organization. 2013.
- Chintamunnee V, Mahomoodally MF. Herbal medicine commonly used against non-communicable diseases in the tropical island of Mauritius. J Herb Med. 2012;2:113-125.
Crossref - Gurib-Fakim A. Medicinal plants: traditions of yesterday and drugs of tomorrow. Mol aspects of Med. 2006;27(1):1-93.
Crossref - Lin LL, Shan JJ, Xie T, et al. Application of traditional Chinese medical herbs in prevention and treatment of respiratory syncytial virus. Evid. Based Complement. Alternat. Med. 2016;6082729.
Crossref - Lin LT, Hsu WC, Lin CC. Antiviral natural products and herbal medicines. J Tradit Complement Med. 2014;4(1):24-35. PMID: 24872930; PMCID: PMC4032839.
Crossref - Dhama K, Karthik K, Khandia R, et al. Medicinal and therapeutic potential of herbs and plant metabolites/extracts countering viral pathogens – Current knowledge and future prospects. Curr Drug Metab. 2018;19(3):236-263.
Crossref - Cheng HM, Li CC, Chen CY, et al. Application of bioactivity database of Chinese herbal medicine on the therapeutic prediction, drug development, and safety evaluation. J. Ethnopharmacol. 2010;132:429-437.
Crossref - Singh PK, Rawat P. Evolving herbal formulations in management of dengue fever. J. Ayurveda Integr. Med. 2017;8:207-210.
Crossref - Chan JF-W, Kok K-H, Zhu Z, et al. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 2020;9:221-236.
Crossref - King AM, Lefkowitz EJ, Mushegian AR, et al. Changes to taxonomy and the International Code of Virus Classification and Nomenclature ratified by the International Committee on Taxonomy of Viruses. Archives of Virol. 2018;163(9):2601-31.
Crossref - Cascella M, Rajnik M, CuomoA, Dulebohn SC, Di Napoli R. Features, evaluation and treatment coronavirus (COVID-19). In: Stat Pearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2020. https://www.ncbi.nlm.nih.gov/books/NBK554776/
- Avula A, Nalleballe K, Narula N, et al. COVID-19 presenting as stroke. Brain Behav. Immun. 2020; in press.
Crossref - Chen N, Zhou M, Dong X, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. 2020;395:507-513.
Crossref - Bukreyev A, Lamirande EW, Buchholz UJ, et al. Mucosal immunisation of African green monkeys (Cercopithecusaethiops) with an attenuated parainfluenza virus expressing the SARS coronavirus spike protein for the prevention of SARS. The Lancet. 2004;363(9427):2122-7.
Crossref - Yang Z-Y, Kong W-P, Huang Y, et al. A DNA vaccine induces SARS coronavirus neutralization and protective immunity in mice. Nature. 2004;428:561-564.
Crossref - Mehmood MA, Sehar U, Ahmad N. Use of bioinformatics tools in different spheres of life sciences. J. Data Mining Genomics Proteomics. 2014;5(2):1.
Crossref - Ferreira LG, Dos Santos RN, Oliva G, Andricopulo AD. Molecular docking and structure-based drug design strategies. Molecules. 2015;20:13384-13421.
Crossref - Sayle RA, Milner-White EJ. RASMOL: biomolecular graphics for all. Trends Biochem. Sci. 1995;20:374-376.
Crossref - Kim S, Chen J, Cheng T, et al. PubChem 2019 update: improved access to chemical data. Nucleic Acids Res. 2019;47:D1102-D1109.
Crossref - O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open Babel: an open chemical toolbox. J. Cheminformatics. 2011;3:33.
Crossref - Hsu K-C, Chen Y-F, Lin S-R, Yang J-M. iGEMDOCK: a graphical environment of enhancing GEMDOCK using pharmacological interactions and post-screening analysis. BMC Bioinform. 2011;12:S33.
Crossref - Babcock GJ, Esshaki DJ, Thomas WD Jr., Ambrosino DM. Amino acids 270 to 510 of the severe acute respiratory syndrome coronavirus spike protein are required for interaction with receptor. J.Virol. 2004;78:4552-4560.
Crossref - Nakayama M, Suzuki K, Toda M, Okubo S, Hara Y, Shimamura T. Inhibition of the infectivity of influenza virus by tea polyphenols. Antivir. Res. 1993;21:289–299.
Crossref - Carneiro BM, Batista MN, Braga ACS, Nogueira ML, Rahal P. The green tea molecule EGCG inhibits Zika virus entry. Virology. 2016;496:215–218.
Crossref - Li J, Song D, Wang S, Dai Y, Zhou J,Gu J. Antiviral effect of epigallocatechingallate via impairing porcine circovirus type 2 attachment to host cell receptor. Viruses. 2020;12:76.
Crossref - Rane JS, Chatterjee A, Kumar A, Ray S. Targeting SARS-CoV-2 spike protein of COVID-19 with naturally occurring phytochemicals: an in silico study for drug development. ChemRxiv. 2020; preprint.
Crossref - Ahmed N, Laverick L, Sammons J, Zhang H, Maslin DJ, Hassan HT. Ajoene, a garlic-derived natural compound, enhances chemotherapy-induced apoptosis in human myeloid leukaemia CD34-positive resistant cells. Anticancer Research. 2001;21:3519-23.
- Ankri S and Mirelman D. Antimicrobial properties of allicin from garlic. Microbes and Infection. 1999;1:125-129.
Crossref - Lin CW, Wu CF, Hsiao NW, et al. Aloe-emodin is an interferon-inducing agent with antiviral activity against Japanese encephalitis virus and enterovirus 71. International Journal of Antimicrobial Agents. 2008;32:355-9.
Crossref - Li SW, Yang TC, Lai CC, et al. Antiviral activity of aloe-emodin against influenza A virus via galectin-3 up-regulation. European Journal of Pharmacology. 2014;738:125-32.
Crossref - Ginwala R, Bhavsar R, Chigbu DG, Jain P, Khan ZK. Potential role of flavonoids in treating chronic inflammatory diseases with a special focus on the anti-inflammatory activity of apigenin. Antioxidants. 2019;8:35.
Crossref - Yusuf MA, Singh BN, Sudheer S, et al. Chrysophanol: a natural anthraquinone with multifaceted biotherapeutic potential. Biomolecules. 2019;9:68.
Crossref - Hewlings SJ, Kalman DS. Curcumin: a review of its’ effects on human health. Foods. 2017;6:92.
Crossref - Seki T, Hosono T, Hosono-Fukao T, et al. Anticancer effects of diallyltrisulfide derived from garlic. Asia Pac J Clin Nutr. 2008;17.
- Lee NH, Lee SM, Song DH, Yang JY, Lee HS. Antimicrobial effect of emodin isolated from Cassia tora Linn. seeds against food-borne bacteria. Journal of Applied Biological Chemistry. 2013;56:187-9.
Crossref - Nikoo M, Regenstein JM, Ahmadi Gavlighi H. Antioxidant and Antimicrobial Activities of (-)-Epigallocatechin-3-gallate (EGCG) and its Potential to Preserve the Quality and Safety of Foods. Compr Rev Food Sci Food Saf. 2018;17:732-53.
Crossref - Yusof YA. Gingerol and its role in chronic diseases. In Drug discovery from mother nature. 2016;177-207.
Crossref - Jimenez-Arellanes A, Luna-Herrera J, Cornejo-Garrido J, et al. Ursolic and oleanolic acids as antimicrobial and immunomodulatory compounds for tuberculosis treatment. Bmc Complementary and Alternative Medicine. 2013;13:258.
Crossref - Wozniak L, Skapska S, Marszalek K. Ursolic acid—a pentacyclictriterpenoid with a wide spectrum of pharmacological activities. Molecules. 2015;20:20614-41.
Crossref - Ahmad B, Rehman MU, Amin I, et al. A review on pharmacological properties of zingerone (4-(4-Hydroxy-3-methoxyphenyl)-2-butanone). The Scientific World Journal. 2015.
Crossref - Kandeel M, Kitade Y. Computational analysis of siRNA recognition by the Ago2 PAZ domain and identification of the determinants of RNA-induced gene silencing. PloS One. 2013;8.
Crossref - Kumar NA, Sharmila R, Akila K, et al. In-silico approach for the assessment of oral cancer property on Limoniaacidissima. Int. J Pharm Sci Rev Res. 2016;7:1271.
- O Trott, AJ Olson, AutoDockVina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading, J Comput Chem.. 2010;31:455-461.
Crossref - DeLano WL. Pymol: An open-source molecular graphics tool. CCP4 Newsletter on protein crystallography. 2002;40:82-92.(Trial version).
- Calligari P, Bobone S, Ricci G and Bocedi A. Molecular Investigation of SARS–CoV-2 Proteins and Their Interactions with Antiviral Drugs. Viruse. 2020;4:445.
Crossref - Ayres JS. Surviving COVID-19: A disease tolerance perspective. Science Advances. 2020;6:18.
Crossref - Min S, Yan M, Kim SB et al. Green Tea Epigallocatechin-3-Gallate Suppresses Autoimmune Arthritis Through Indoleamine-2,3-Dioxygenase Expressing Dendritic Cells and the Nuclear Factor, Erythroid 2-Like 2 Antioxidant Pathway. J Inflamm. 2015;12:53.
Crossref - Saeed M, Naveed M, Arif M, et al. Green tea (Camellia sinensis) and l-theanine: Medicinal values and beneficial applications in humans-A comprehensive review. Biomed Pharmacother. 2017;95:1260-1275.
Crossref
© The Author(s) 2020. Open Access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License which permits unrestricted use, sharing, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.