


ISSN: 0973-7510
E-ISSN: 2581-690X
India is well known for its resources of medicinal plants and their application in different diseases. Though costly, herbal drugs may be used in addition with synthetic drugs to increase the effectivity of the therapy. In 21st century the world had experienced the pandemic of SARS-CoV-2. The battle is still on against this deadly virus as more variants are yet to come. Though we have developed some immunity, still researches are going on to combat the viruses with newer combinations of drugs with less side effects. Moreover, targeting a conserved essential protein of the virus with a drug is more acceptable to the community of medical practitioners, as it will minimize the range of drugs to be administered against SARS-CoV-2. In this study, we have selected the delta variant of the virus due to its most detrimental record on human health. We have tried to establish the structural similarity of RNA dependent RNA polymerase of SARS-CoV-2 delta variant with the wild type one and then established the in silico interaction of bioactives from ginger on the ligand binding pockets of concerned protein, as a first step to design herbal drugs against this deadly virus.
Homology Modelling, Herbal Drug, Medicinal Plant, [6]-Dehydrogingerdione, Gingerenone
SARS-CoV-2 has a zoonotic origin and there are evidences of both human-to-human and human-to-animal transmission with an ongoing evolution that can be tracked through GISAID.1,2 On 10th Jan 2020, its very first genome sequence (WH-Human_1) was announced by China. On 23rd Feb, Wuhan, declared the news of an emerging novel coronavirus all over the world.3 The global pandemic of SARS-CoV-2 infection increased its mortality rate during the outburst of its delta variant and the real cause of panic among the people was the lack of treatment throughout the world. Though the target of vaccine and most of the drug development was attached to target the S protein,4 which is essential in host interaction and viral fusion,5 but the RNA dependent RNA Polymerase (RNA dependent RNA polymerase) of the virus, is another major target for drug development against SARS-CoV-2. RNA dependent RNA polymerase is the key protein required for viral genome multiplication and if blocked, the propagation of virus have discontinued.6
The RNA dependent RNA polymerase of SARS-CoV-2 is a product of viral ORF1ab gene that codes for around sixteen non-structural viral replicase polyprotein (nsp),7,8 which undergoes post-translational cleavage to give functional RNA dependent RNA polymerase (nsp 12) by 3C like protease (nsp 5) and Papain like protease (nsp 3).9,10 Screening of RNA dependent RNA polymerase from various RNA virus family have shown that the molecular weight of the protein ranges from 240-450 Dalton with structural similarity with human right hand having a clear palm, finger and thumb domain.11,12
The sequential identity and structural similarity between the RNA dependent RNA polymerase of SARS-CoV and SARS-CoV-2 is well documented.13 Keeping faith on the structural homology, RNA dependent RNA polymerase inhibitor antiviral drugs administered against SARS-CoV-2 have gained their importance to the medical practitioners for treating the SARS-CoV-2 patients also.14 Different synthetic nucleoside analogues namely Sofosbuvir, Remdisivir, Ribavirin etc have been documented for their successful binding with the active site of RNA dependent RNA Polymerase of SARS-CoV-2,15 however, for the recent pandemic, only one RNA dependent RNA polymerase inhibitor antiviral drug, Remdesivir, has been sanctioned by United States Food and Drug Administration authority for using to the patients having severe SARS-CoV-2 infection.15 Remdesivir is injected intravenously to the
SARS-CoV-2 infected patients with age ≥12 years and body weight ≥40 kgs, depending upon the severity of the symptoms. However, due to scarcity of supply and high cost of Remdesivir during the outbreak of delta variant of SARS-CoV-2 in India, it became unaffordable to many people.
India, being a land of Ayurveda, people here are well acquainted with the use of different plant extract for treating different diseases, specifically the seasonal infections. For treating common cold, sore-throat like symptoms, use of extracts from Ginger, Garlic, Tulsi is largely accepted.16 Effect of different Himalayan medicinal plant extracts and their binding potential with RNA dependent RNA polymerase of SARS-CoV-2 has been reported earlier.17 However, there exact mode of actions against SARS-CoV-2 has not been reported earlier. However, different phytochemicals from Himalayan medicinal plants have been docked successfully with SARS-CoV-2 RNA dependent RNA polymerase protein. In a very recent study, 271 phytochemicals from 25 Himalayan plants have been successfully screened against 9 SARS-CoV-2 proteins like RNA dependent RNA polymerase, viral protease, helicase, envelop and capsid protein.18 In this background, we have done an in silico analysis to determine whether the biomolecules present in the common medicinal plants like ginger can successfully bind with the RNA dependent RNA polymerase of SARS-CoV-2 delta variant or not.
Retrieval of RNA dependent RNA polymerase protein sequence of SARS-CoV-2 wild type and delta variant and Alignment for comparison
The sequence of wild type and delta (B.1.617.2) variants of SARS-CoV-2 have been obtained from NCBI GenBank against the accession numbers NC_045512.2 and MZ208926 respectively.19 The RNA dependent RNA polymerase gene sequence was retrieved from the whole genome sequences with aforesaid accession number and converted to protein using tBLASTx.20 Both the RNA dependent RNA polymerase protein sequences were aligned with Clustal Omega21 to determine position specific changes in amino acids.
Prediction of secondary structure of RNA dependent RNA polymerase protein sequence of SARS-CoV-2 wild type and delta variant
Secondary structures of both the RNA dependent RNA polymerase proteins were predicted using PSIPRED workbench22 in PSIPRED server23 and their relative comparison was done. Disorder prediction within the structures was done using DISOPRED3 server,24 TM topology and Helix packing was established with MEMPACK server25 and the domain folds were recognised using pDomTHREADER.26,27 Probable domains present in the structures were predicted using DomPred.28
Homology modelling
Homology modelling was performed to determine the quaternary structure using SWISS MODEL,29 IntFOLD and RaptorX server.
SWISS MODEL is a web-based application used for automated homology modelling, accessible through ExPasy web server. The sequences of interest, both the wild type and Delta variant RNA dependent RNA polymerase were subjected to 3D structure analysis. Using protein 7krn.1A as template from SWISS MODEL repository,30 both the protein structures were modelled with the help of pairwise alignment. The quality of the model was assessed by calculating the reliability of the resulting models based on the GMQE and QMEAN score.31 GMQE score is used to find out the most suitable template as it denotes the most likely structural similarity between the template and the query.32 The QMEAN score measures the degree of nativeness between the template and the query and is being normalized on the number of interactions.33 The relative stability of the model was assigned through Ramachandran plot. The predicted model was further validated using RaptorX.34
More intrinsic study regarding the structure prediction was performed using IntFOLD35 where model quality assessment was done using ModFOLD5, intrinsic disorders were predicted using DISOclust3, total number of different domains along with their individual position were predicted using DomFOLD3, available and probable function and ligand binding sites were determined FunFOLD3. Model quality with respect to optimized hydrogen atom placement and atomic contact analysis was assessed using MolProbity score.36
Prediction of active site residues of RNA dependent RNA polymerase of SARS-CoV-2 delta variant
The server PROSITE37 is the first annotated collection of motif descriptors for the recognition of protein families and domains. SARS-CoV-2 delta variant RNA dependent RNA polymerase sequence (MZ208926) was submitted as input sequence in PROSITE to find out the active site domain and more specifically the residues involved in substrate binding.
Determination of available ligand binding pocket in RNA dependent RNA polymerase of SARS-CoV-2 delta variant
DoGSiteScorer38 webserver was used to predict the available ligand binding pockets present on the RNA dependent RNA polymerase protein of SARS-CoV-2 Delta variant. It is an easy-to-use webserver to predict pockets and sub-pockets of a protein structure of interest.
Determination of drug likeliness of selected bioactives from Ginger (Zingiber officinale)
Information regarding the major bioactives present in Ginger was obtained from Arcusa et al.39 Major bioactives were selected based on their relative proportion in the corresponding extract. Structures (in SDF format) of these selected bioactives were derived from PubChem.40 Drug likeliness of the compounds were determined with reference to Lipinski’s rule of five using DruLiTo, developed and maintained by Department of Pharmacoinformatics, NIPER, India. Bioactives that fails at least one parameter as per Lipinski’s rule of five had not been considered for further analysis.41 Rest of the bioactives were analysed for refractivity and Number of atoms through CMC50 like rule and QED using DruLiTo.
Here also, Remdesivir was used as positive control. Those who qualified these parameters, are subjected to study for their ADMET properties by admetSAR.42
Docking of PubChem derived structures of selected bio actives of ginger with RNA Dependent RNA Polymerase of SARS-CoV-2 Delta variant
Structures of bioactives those qualify the drug likeliness were first converted to PDB file by Online SMILES translator maintained by The CADD Group’s Chemoinformatics Tools and User Services. Bioactives that qualified through Lipinski’s rule, CMC 50 like rule, QED and have favorable ADMET properties were docked against the protein of interest variant by using the webserver PatchDock.43 PDB file of both the target protein (SARS-CoV-2 RNA dependent RNA polymerase) and the bioactive compounds were submitted as an input and analysed with clustering RMSD value of 1.5 and small ligand protein as its complex type.
Out of all the docked structures the structures with minimum ACE values (lowest 10) were taken into considerations and corresponding PDB files were retrieved. The docked structure was viewed using UCSF ChimeraX (or simply ChimeraX)44,45 and the binding site was compared along with the data available from DoGSiteScorer analysis. The widely used drug Remdesivir was used as reference in docking analysis.
The binding of Remdesivir has been further compared and confirmed with that of [6]-Dehydrogingerdione and Gingerenone A by analysis through AutoDock.46 Binding affinity of each docking results were analyzed and lowest binding affinity and RMSD value has been considered.
Detection of alteration in amino acid sequence in RNA dependent RNA polymerase of SARS-CoV-2 delta variant with that of the wild type
Clustal omega analysis of the two converted amino acid sequences retrieved from NCBI database showed that there is a difference in only three amino acids in the RNA dependent RNA polymerase of SARS-CoV-2 delta variant with compare to the wild type. In position 228 and 671 Glycine of the wild type variant has been converted into Serine whereas in position 323, a Proline residue present in the wild type variant has been altered to Leucine (Supplementary Figure 1).
Prediction of secondary structure of RNA dependent RNA polymerase of SARS-CoV-2 wild type and delta variant
PSIPRED prediction of secondary structure of the both 932 residue protein shows highly conserved secondary structural distribution throughout the whole protein. Both the protein consists of 38 alpha helixes and 30 beta strands distributed throughout the whole protein including one putative domain boundary almost at the same position (at position 370 in case of the wild type RNA dependent RNA polymerase and at position 367 in case of the Delta variant RNA dependent RNA polymerase). Rest part of both the proteins are composed of random coil which suggests same type of overall folding in both the proteins (Supplementary Table 1).
As per PSIPRED prediction, it was also evident that around 22.75% hydrophobic residues are distributed throughout both the entire wild type and delta variant RNA dependent RNA polymerase protein without any large hydrophobic patches, suggests that there is no membrane spanning region in the protein. This observation is also supported by the secondary structure analysis data from PSIPRED that shows absence of any transmembrane helix, region of membrane interaction and any membrane directed signal peptides (Supplementary Figure 2).
Homology modelling with SWISS MODEL, IntFold and RaptorX
SWISS MODEL analysis of both the protein against 7krn.1A shows around 99.68% sequence identity with 100% coverage during homology modelling. As both the proteins are present as monomer, so QSQE score was not detected. The GMQE score of 0.91 for the wild type protein with QMEANDisCo score 0.89 ± 0.05 and GMQE 0.90 with QMEANDisCo score 0.88 ± 0.05 for the delta type variant confirms the overall improved model quality. When Ramachandran plot was analysed for determining the stability of the structure, it shows that for both the protein around 94% residues lied in the favourable region and only around 4% are in the outliers. However, the more detailed analysis of the same shows that the serine in position 671 of the delta variant occupies almost outer boundary of Ramachandran formidable region that may questions the stability of this particular torsion angles. On the other hand, the other two mutations located at position 228 and 323 are however lies in the same quadrant of the plot with slightly altered value of corresponding torsion angles, hence may not affect the stability of the protein (Figure 1).
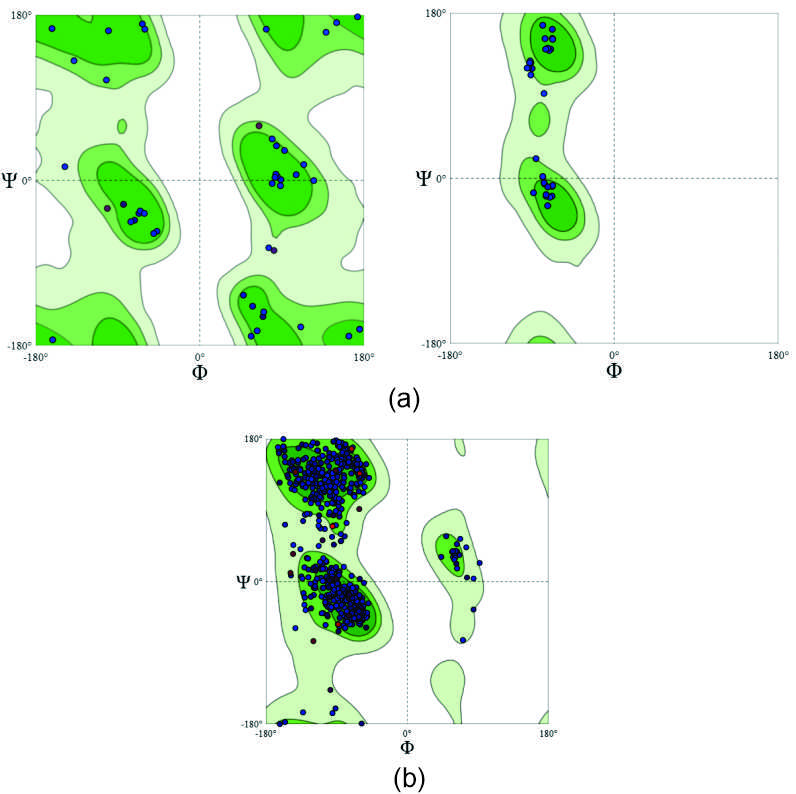
Figure 1. Ramachandran Plot shows the Ф and Ж distributions of non-glycine, non-proline residues and give residues distribution. The Ф and Ж angles originated were plotted against each other to differentiate the favourable and non-favourable regions. These angles were used to evaluate the quality of regions. (a) Denotes glycine at 228 and 671 and proline at 323 of SARS-CoV-2 wild type and (b) Represents sequence diversion of SARS-CoV-2 delta variant from wild type i.e., Serine, leucine, serine
C-beta deviation values from Ramachandran plot using RaptorX shows that both wild type and delta variant RNA dependent RNA polymerase shares almost same structural adaptation in all the C-beta deviation points, however, the mutant protein lacks an additional deviation point at position 377 (Supplementary Table 2).
The information regarding the binding of ligands with their probable binding site in the predicted 3D structure has been obtained from the sub components of IntFold analysis. The FunFOLD score was found as 0.64 in wild type protein and 0.53 in mutant one having equal number of binding sites in both the proteins. Not only the number but also the position of ligand bindings was same (at position 295, 301, 306, 310,487, 642, 645, 646), except one where position 209 of wild type protein has been replaced by position 218 in mutant type. The disorder prediction curve from IntFold analysis also predicts a highly ordered structure from position 200 to 800 of both the protein (Supplementary Figure 3). The overall modelled structure for both the proteins are represented in Figure 2. No such overall distortion in the structure of the wild type protein has been observed due to the mutation and there is no significant structural differences between these two proteins.
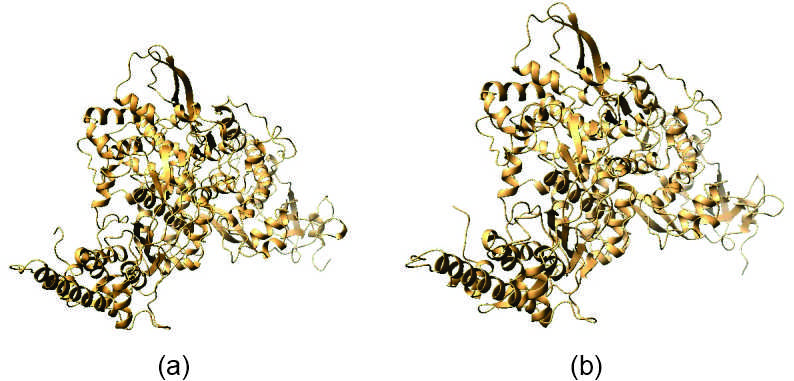
Figure 2. 2D structures from IntFOLD. (a) SARS-CoV-2 wild type and (b) SARS-CoV-2 delta variant
Detection of active site residue and ligand binding pockets in RNA dependent RNA polymerase of SARS-CoV-2 delta variant
The PROSITE data shows that both the protein has RNA dependent RNA polymerase activity and the pivotal active site residues lies within the region spanning from 759 to 761 which consists of one serine followed by two consecutive aspartic acids (Supplementary Figure 4).
Total 44 ligand binding sub-pockets are distributed among 35 major pockets unequally, as found in DOGSITE scorer analysis (Supplementary Table 3). Among them pocket P_2 has the highest drugscore (0.81003) while P_25 has the lowest one (0.19103). Thus it can be said that binding of any ligand at P_2 increases its probability to act as a drug after further clinical trial.
Determination of drug likeliness of selected bioactives Ginger (Zingiber officinale)
As per Lipinski’s rule, the molecular weight of Remdesivir was found 602.23, the lipophilicity (log P) 0.336, H-bond donor 4, H-bond acceptor. The molecular weight of all the bioactives from Ginger were well within the permissible range of druggability (Table 1). Lipophilicity of all the bioactives from the Ginger is quite higher and five of them, namely Alpha Cucurmene (5.761), Alpha Farnesene (6.136), Beta Bisabolene (5.551), Beta Sesquiphellandrene (6.399) and Zingiberene (6.354) exceeds the permissible limit also (Table 1). Number of H bond donor and acceptor atom present in the bioactives of Ginger are less than five (Table 1).
Table (1):
Analysis of drug likeliness of bioactives of Zingiber officinale through Lipinski’s rule
Bioactives |
Title |
MW |
logP |
HBA |
HBD |
|---|---|---|---|---|---|
[6]-Dehydrogingerdione |
22321203 |
290.15 |
3.193 |
4 |
2 |
6-Gingerol |
442793 |
294.18 |
2.437 |
4 |
2 |
8-Gingerol |
168114 |
322.21 |
3.575 |
4 |
2 |
10-Gingerol |
168115 |
350.25 |
4.713 |
4 |
2 |
Alpha Curcumene |
92139 |
202.17 |
5.761 |
0 |
0 |
Alpha Farnesene |
5281516 |
204.19 |
6.136 |
0 |
0 |
Beta Bisabolene |
10104370 |
204.19 |
5.551 |
0 |
0 |
Beta Sesquiphellandrene |
519764 |
204.19 |
6.399 |
0 |
0 |
Gingerenone A |
5281775 |
356.16 |
2.421 |
5 |
2 |
Paradol |
94378 |
278.19 |
3.889 |
3 |
1 |
Shogaol |
5281794 |
276.17 |
3.775 |
3 |
1 |
Zingerone |
31211 |
194.09 |
0.791 |
3 |
1 |
Zingiberene |
92776 |
204.19 |
6.354 |
0 |
0 |
To qualify, MW (molecular weight) should be <=500 Dalton, HBA (Hydrogen bond acceptor) <=10, HBD (Hydrogen bond donor) <=5 and LogP <= 5 for respective bioactives
The bioactives that qualifies Lipinski,s rule of five, were also able to qualify through CMC 50 like rule except 10-Gingerol and Gingerone (Table 2). Remdesivir, the widely used drug against SARS-CoV-2 also does not qualify the CMC 50 like rule. All the bioactives except 10-Gingerol of Ginger have both weighted and unweighted QED value greater than 0.5 (Table 3). Bioactives from Ginger except shogaol qualifies for their ADMET properties
(Table 4).
Table (2):
Analysis of drug likeliness of bioactives of Zingiber officinale through CMC-50 like rule
Bioactives |
AMR |
nAtom |
MW |
LogP |
|---|---|---|---|---|
[6]-Dehydrogingerdione |
84.68 |
43 |
290.15 |
3.193 |
6-Gingerol |
80.8 |
47 |
294.18 |
2.437 |
8-Gingerol |
86.63 |
53 |
322.21 |
3.575 |
10-Gingerol |
92.45 |
59 |
350.25 |
4.713 |
Gingerenone A |
110.29 |
50 |
356.16 |
2.421 |
Paradol |
77.75 |
46 |
278.19 |
3.889 |
Shogaol |
82.07 |
44 |
276.17 |
3.775 |
Zingerone |
58.17 |
28 |
194.09 |
0.791 |
Alpha Curcumene |
71.56 |
37 |
202.17 |
5.761 |
Alpha Farnesene |
73.63 |
39 |
204.19 |
6.136 |
Beta Bisabolene |
69.93 |
39 |
204.19 |
5.551 |
Beta Sesquiphellandrene |
68.91 |
39 |
204.19 |
6.399 |
Zingiberene |
70.13 |
39 |
204.19 |
6.354 |
To qualify, AMR (Atomic refractivity) must be present in between 70 to 110, nAtom or atom count 30 to 55, LogP must fall in between 1.3 to 4.1 and their respective MW should be present in between 230 to 390
Table (3):
Analysis of drug likeliness of bioactives of Zingiber officinale through weighed and unweighed QED
Bioactives |
TPSA |
nRB |
SAlerts |
nAroma Ring |
uwQED |
wQED |
|---|---|---|---|---|---|---|
[6]-Dehydrogingerdione |
66.76 |
8 |
4 |
1 |
0.597 |
0.425 |
6-Gingerol |
66.76 |
10 |
1 |
1 |
0.693 |
0.643 |
8-Gingerol |
66.76 |
12 |
1 |
1 |
0.59 |
0.537 |
10-Gingerol |
66.76 |
14 |
1 |
1 |
0.486 |
0.431 |
Gingerenone A |
75.99 |
9 |
1 |
2 |
0.794 |
0.753 |
Paradol |
46.53 |
10 |
1 |
1 |
0.718 |
0.67 |
Shogaol |
46.53 |
9 |
2 |
1 |
0.756 |
0.675 |
Zingerone |
46.53 |
4 |
0 |
1 |
0.834 |
0.801 |
Alpha Curcumene |
0.0 |
4 |
1 |
1 |
0.307 |
0.494 |
Alpha Farnesene |
0.0 |
6 |
2 |
0 |
0.233 |
0.355 |
Beta Bisabolene |
0.0 |
4 |
1 |
0 |
0.323 |
0.517 |
Beta Sesquiphellandrene |
0.0 |
4 |
1 |
0 |
0.336 |
0.538 |
Zingiberene |
0.0 |
4 |
1 |
0 |
0.325 |
0.521 |
To qualify, unQED must be >=0.5 and wQED must be >=0.5 for respective bioactives
Table (4):
Assessment of environment and human hazard properties of bioactives of Zingiber officinale was checked through admetSAR
| Bioactive | [6]- Dehydrogingerdione | 6-Gingerol | 8-Gingerol | 10-Gingerol | Gingerenone A | Paradol | Shogaol | Zingerone | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Human intestinal absorption | + | 0.9853 | + | 0.9924 | + | 0.9924 | + | 0.9924 | + | 0.9839 | + | 0.9928 | + | 0.9904 | + | 0.9928 |
| Caco-2 permeability | + | 0.7089 | + | 0.5943 | + | 0.6241 | – | 0.5240 | – | 0.5893 | + | 0.8109 | + | 0.7765 | + | 0.7782 |
| Organic cation transporter protein 2 inhibitor | – | 0.7814 | – | 0.8000 | – | 0.8000 | – | 0.8000 | – | 0.9000 | – | 0.7750 | – | 0.7750 | – | 0.9500 |
| P-gp inhibitor | – | 0.8931 | – | 0.8934 | – | 0.8535 | – | 0.7414 | + | 0.6603 | – | 0.9078 | – | 0.8852 | – | 0.9849 |
| P-gp substrate | – | 0.6973 | + | 0.5562 | + | 0.5663 | + | 0.5663 | – | 0.8812 | – | 0.6228 | – | 0.7294 | – | 0.8638 |
| CYP3A4 substrate | – | 0.5352 | + | 0.5566 | + | 0.5690 | + | 0.5690 | – | 0.5000 | – | 0.5000 | + | 0.5317 | – | 0.5832 |
| CYP2C9 substrate | – | 0.7991 | – | 0.7974 | – | 0.7974 | – | 0.7974 | – | 0.7936 | – | 0.5963 | – | 0.7929 | – | 0.5963 |
| CYP2D6 substrate | – | 0.7911 | + | 0.3792 | + | 0.3792 | + | 0.3792 | – | 0.7873 | + | 0.3653 | – | 0.8137 | + | 0.3653 |
| CYP3A4 inhibitor | – | 0.6157 | – | 0.5902 | – | 0.5902 | – | 0.5902 | – | 0.5395 | – | 0.8195 | – | 0.7130 | – | 0.9322 |
| CYP2C9 inhibitor | – | 0.9128 | – | 0.8278 | – | 0.8278 | – | 0.8278 | + | 0.5938 | – | 0.8987 | – | 0.9117 | – | 0.9274 |
| CYP2C19 inhibitor | + | 0.5681 | – | 0.6350 | – | 0.6350 | – | 0.6350 | + | 0.8534 | – | 0.6454 | – | 0.5831 | – | 0.7111 |
| CYP2D6 inhibitor | – | 0.8270 | – | 0.7926 | – | 0.7926 | – | 0.7926 | – | 0.7605 | – | 0.8405 | – | 0.8292 | – | 0.8489 |
| CYP1A2 inhibitor | + | 0.5869 | + | 0.6632 | + | 0.6632 | + | 0.6632 | + | 0.8823 | + | 0.5314 | + | 0.6002 | + | 0.6701 |
| CYP inhibitory promiscuity | – | 0.8503 | – | 0.8581 | – | 0.8581 | – | 0.8581 | + | 0.6359 | – | 0.8421 | – | 0.7155 | – | 0.8882 |
| Carcinogenicity (binary) | – | 0.7731 | – | 0.7000 | – | 0.7000 | – | 0.7000 | – | 0.8073 | – | 0.6571 | – | 0.7731 | – | 0.6571 |
| Carcinogenicity (trinary) | NR | 0.7234 | NR | 0.7188 | NR | 0.7188 | NR | 0.7188 | NR | 0.7289 | NR | 0.6791 | NR | 0.6917 | NR | 0.6489 |
| Ames Mutagenecity | – | 0.7600 | – | 0.5700 | – | 0.6100 | – | 0.6100 | – | 0.6100 | – | 0.5700 | – | 0.7400 | – | 0.7200 |
| hERG inhibitor | – | 0.3680 | + | 0.7369 | + | 0.6726 | + | 0.7163 | – | 0.4865 | + | 0.7523 | – | 0.4031 | – | 0.5789 |
| Acute Oral Toxicity | III | 0.6401 | III | 0.6007 | III | 0.6007 | III | 0.6007 | III | 0.6974 | III | 0.8126 | III | 0.6916 | III | 0.8801 |
Where, NR indicates not required, sign (+/-) and number represents value and probability respectively.
Docking of druggable bioactives with RNA dependent RNA polymerase of SARS-CoV-2 delta variant
PatchDock software approaches a method to compute the atomic contact energy of ligand and receptor. Out of all, [6]-Dehydrogingerdione and Gingerenone A of Ginger are successfully screened through admetSAR, Lipinski’s Rule, CMC 50 like rule and both weighted and unweighted QED values. Further they are docked against SARS-CoV-2 Delta variant by retrieving PDB structures from PubChem Database. It is seen that the lowest ACE values -341.75 and -411.27 for [6]-Dehydrogingerdione and Gingerenone A, respectively, provide insight into the most stable docked conformations.
Further visualizing their respective receptor-ligand interactions through Chimera X (Figure 3a and 3b) and overlapping (Figure 3a and 3b) them with respect to already discovered effective drug Remdesivir, to check whether the docking has happened in the same pocket for both cases through Overlay Images Online-PineTools.
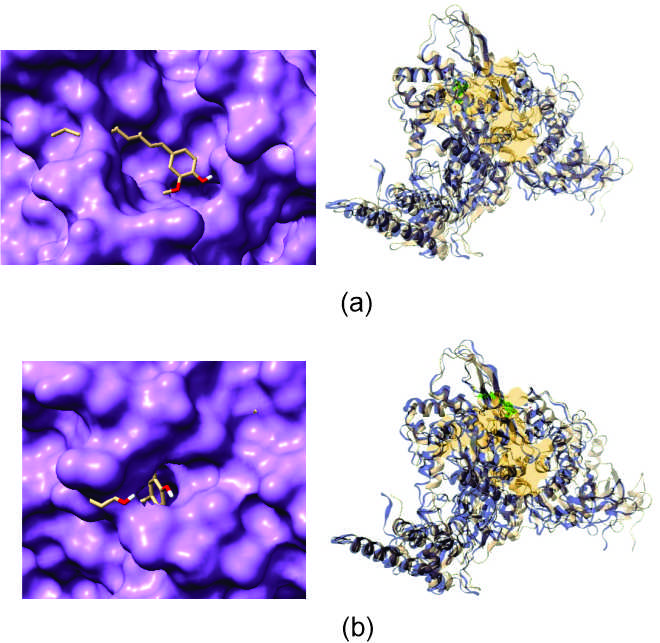
Figure 3. Docked structures of lowest ACE valued structure (a). [6]-Dehydrogingerdione and (b). Gingerenone A with RNA dependent RNA polymerase of SARS-CoV-2 delta variant through PatchDock
Remdesivir binds with RNA dependent RNA polymerase protein of SARS-CoV-2 delta variant in the ligand binding pocket, P_0. It is observed that the screened bio-actives binds to that same ligand binding pocket, P_0 of RNA dependent RNA polymerase protein of SARS-CoV-2 delta variant with certain interaction of amino acids (THR 324, SER 325, SER 384, PHE 326, GLY 327, PRO 328, LEU 329, VAL 330, VAL 398, VAL 341, MET 666, MET 380, ALA 382, ALA 383, ALA 379, HIS 381) for [6]-Dehydrogingerdione and (LEU 648, LEU 302, PHE 652, SER 649, ASN 300, VAL 299, CYS 301, CYS 298, CYS 306, ASP 304, ASP 303, ILE 307, ARG 651, ARG 305) Gingerenone A respectively (Table 5, Figure 4).
Table (5):
Overlapped structures with lowest ACE value with respect to SARS-CoV-2 delta variant through Overlay Images Online-PineTools and their respective interactions with SARS-CoV-2 Delta Variant
Name of the bioactives |
ACE value |
Binding |
Pockets |
|---|---|---|---|
[6]-Dehydrogingerdione |
-341.75 |
THR 324, SER 325, SER 384, PHE 326, GLY 327, PRO 328, LEU 329, VAL 330, VAL 398, VAL 341, MET 666, MET 380, ALA 382, ALA 383, ALA 379, HIS 381 |
P_0 |
Gingerenone A |
-411.27 |
LEU 648, LEU 302, PHE 652, SER 649, ASN 300, VAL 299, CYS 301, CYS 298, CYS 306, ASP 304, ASP 303, ILE 307, ARG 651, ARG 305 |
P_0 |
Remdesivir |
-586.5 |
VAL 341, VAL 330, VAL 398, ALA 379, ALA 399, ALA 382, ALA 383, PRO 378, PRO 328, LEU 329, LEU 323, LEU 388, LEU 389, LEU 387, GLY 327, MET 666, PHE 326, PHE 396, SER 325, SER 397, THR 324, ASP 390 |
P_0 |
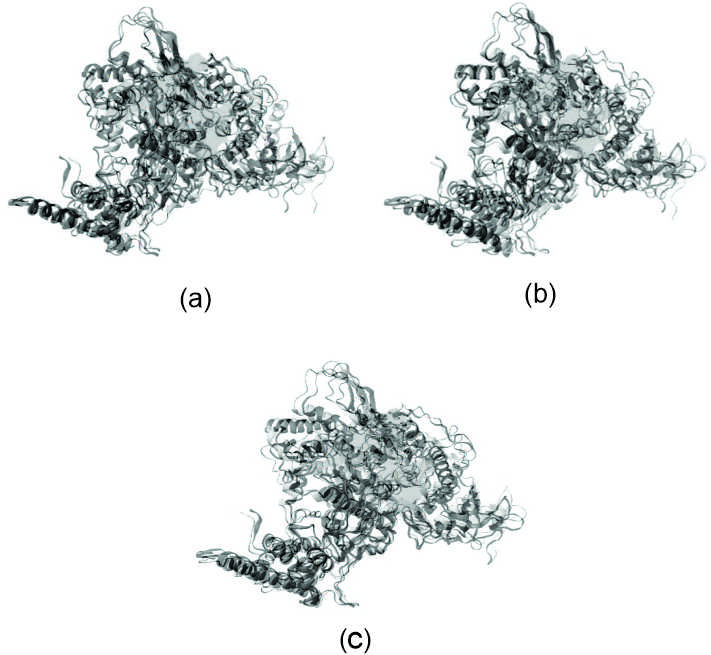
Figure 4. Overlapping structures with respect to SARS-CoV-2 Delta variant. (a) [6]-Dehydrogingerdione overlapped with SARS-CoV-2 Delta Variant; (b) Gingerenone A overlapped with SARS-CoV-2 Delta Variant; (c) Remdesivir overlapped with SARS-CoV-2 Delta Variant
In docking with AutoDock, [6]-Dehydrogingerdione shows 9 binding site, where docking with highest binding affinity (Red circle in Figure 5a) does not corresponds to the binding site of Remdisivir, however, there also exist another binding site with comparatively lower affinity (Green circle in Figure 5a) that are same with that of Remdisivir. and also corresponds to Pocket P_0 of RNA dependent RNA polymerase. On the other hand, the best possible binding of Gingerenone A (Figure 5b) almost coincides with that of Remdisivir (Figure 5c) at Pocket P_0.
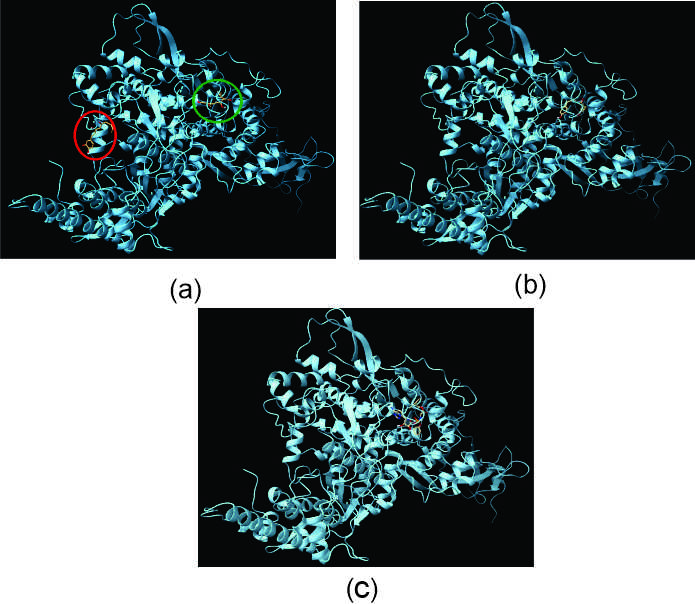
Figure 5. Docked structures of lowest ACE valued structure. (a) [6]-Dehydrogingerdione; (b) Gingerenone A; (c) Remdisivir with RNA dependent RNA polymerase of SARS-CoV-2 delta variant through PatchDock. [6]-Dehydrogingerdione can binds with the protein at possible two sites with different affinity. The green circle resembles the binding site for Remdesivir
The glycine to serine interconversion is a common biological phenomenon47 but the replacement of a non-polar residue (Glycine) with a polar (Serine) one may influence the functionality of an enzyme by modulating the binding and bending ability of the polypeptide chain. However, the conversion of an imino acid (Proline) to a non-polar aliphatic amino acid (Leucine) might definitely have some influence in formation of the secondary structure of the molecule. Though the effect of these changes over the functionality of the enzyme needs to be revealed.
However, if protein folding machinery is concerned, random coil establishes the connections between the ordered secondary structure like helix and beta sheet, and themselves are highly disordered entity.48 If the site of mutations associated with the concerned proteins are taken into account, we have observed that all the changes in amino acid residues are present in the random coil part of the RNA dependent RNA polymerase of delta variant (according to PSIPRED result only). So, it can be assumed that in spite of having mutations, both the proteins do not have remarkable variation in folding pattern that supports the conservation of RNA dependent RNA polymerase structure, however, phenotypically it is proved that Delta variant of the virus has higher rate of replication and multiplication than the wild type.
Hydrophobic amino acids are thought to be the initiator of protein folding and the distribution of hydrophobic amino acids guides the folding pattern of a protein.49 Presence of almost equal number of hydrophobic amino acids at almost same position in both the RNA dependent RNA polymerase variants supports their same type of folding with similar kind of activities, probably guided by same pathway. Moreover, the percentage and distribution of polar and non-polar residues within these two types of RNA dependent RNA polymerase are also almost equivalent. 33.36% polar residue in both the variant of RNA dependent RNA polymerase, whereas non polar residue in wild type and delta variant RNA dependent RNA polymerase is 27.14% and 27.03%, respectively. The slightly higher value of polar residue in the wild type is due to the conversion of two polar glycine of wild type RNA dependent RNA polymerase to non-polar serine residue in delta variant.
Considering the overall conserved structure of RNA dependent RNA polymerase in different SARS-CoV-2 variants, it will it is more logical to design drugs against this deadly disease by targeting RNA dependent RNA polymerase. We may also propose that since RNA dependent RNA polymerase is more or less conserved in major Coronavirus lineages, a proper RNA dependent RNA polymerase inhibitor may allow us to strengthen our therapeutic repertoire against the emerging and re-emerging viral pathogens. Remdesivir, a well-known drug designed against viral RNA dependent RNA polymerase have several side effects,50 which may be fatal in few incidences. In spite of this, Remdisivir, along with other synthetic RNA dependent RNA Polymerase inhibitors have become the primary choice to the medical practitioners due to their more specific action and availability. Common herbal plants are well known for their medicinal activities against common cough and cold and symptomatic treatment with these is an age-old process. Most of the ingredients, if not all, present in a medicinal plant, have some beneficial role in combating different infections. Minimum side effects or toxicity has still been reported against any medicinal plants. Use of Ginger for relaxing from sore throat and other types of chest congestion is a common practice in India.6 dehydrogingerone and Gingerenone A, two major phytochemicals from ginger extract have been documented for binding with 3C like protease of SARS-CoV-2.51 In this study these two components have been proved to bound with a druggable pocket of RNA dependent RNA Polymerase of the same virus also. It can be hypothesised from this point of observation that imposing few modifications in the side chain of these two components by addition and/or deletion of small groups without changing their basic structure may alter the site of binding to a more druggable one. Successful binding of these two components with RNA dependent RNA polymerase of delta variant of SARS-CoV-2 may enhance its probability to be considered as a druggable component against this disease. However, taking the universal identity of SARS-CoV-2 RNA dependent RNA polymerase, it can also be tested against other upcoming variants of this deadly virus.
This report has established the in silico binding of two major druggable components of Ginger, namely [6]-Dehydrogingerdione and Gingerenone A with RNA dependent RNA polymerase of SARS-CoV-2 delta variant. However, the results are preliminary and further extensive studies required to find out the feasibility of the result.
Additional file: Additional Table S1-S3 and Figure S1-S4.
ACKNOWLEDGMENTS
The authors are thankful to Bidhannagar College, Kolkata, India, and St. Xavier’s College, Kolkata, India, for providing facilities to carry out the research.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
AUTHORS’ CONTRIBUTION
TD collected the data and performed the experiments. UDG performed data analysis and wrote the manuscript. Both authors read and approved the final manuscript for publication.
FUNDING
None.
DATA AVAILABILITY
All datasets generated or analyzed during this study are included in the manuscript and/or in the supplementary files and were obtained from NCBI databases.
ETHICS STATEMENT
Not applicable.
- Singh D, Yi SV. On the origin and evolution of SARS-CoV-2. Exp Mol Med. 2021;53(4):537-547.
Crossref - Khare S, Gurry C, Freitas L, et al. GISAID’s role in pandemic response. China CDC Weekly.2021;3(49):1049-1051.
Crossref - Zheng J. SARS-CoV-2: an emerging coronavirus that causes a global threat. Int J Biol Sci. 2020;16(10):1678-1685.
Crossref - Du L, He Y, Zhou Y, Liu S, Zheng BJ, Jiang S. The spike protein of SARS-CoV-a target vaccine and therapeutic development. Nat Rev Microbiol. 2009;7(3):226-236.
Crossref - Muhammed Y, Nadabo AY, Pius M, et al. SARS-CoV-2 spike protein and RNA dependent RNA polymerase as targets for drug and vaccine development: a review. Biosaf Health. 2021;3(5):249-263.
Crossref - Pathania S, Rawal RK, Singh PK. RNA dependent RNA polymerase (RNA-dependent RNA polymerase): a key target providing anti-virals for the management of various viral diseases. J Mol Struct. 2022;1250:131756.
Crossref - Subissi L, Imbert I, Ferron F, et al. SARS-CoV ORF1b-encoded nonstructural proteins 12-16: replicative enzymes as antiviral targets. Antiviral Res. 2014;101:122-130.
Crossref - Gordon DE, Jang GM, Bouhaddou M, et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. 2020;583(7816):459-468.
Crossref - Aftab SO, Ghouri MZ, Masood MU, et al. Analysis of SARS-CoV-2 RNA-dependent RNA polymerase as a potential therapeutic drug target using a computational approach. J Transl Med. 2020;18(1):275-290.
Crossref - Khan MT, Irfan M, Ahsan H, et al. Structures of SARS-CoV-2 RNA-binding proteins and therapeutic targets. Intervirology. 2021;64(2):55-68.
Crossref - Smertina E, Urakova N, Strive T, Frese M. Calicivirus RNA-dependent RNA polymerases: evolution, structure, protein dynamics, and function. Front Microbiol. 2019;10:1280-1302.
Crossref - Hengxia J, Gong P. A structure-function diversity survey of the RNA-dependent RNA polymerases from the positive-strand RNA viruses. Front Microbiol. 2019;10:1945-1956.
Crossref - Wu C, Liu Y, Yang Y, et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm Sin B. 2020;10(5):766-788.
Crossref - Min JS, Kwon S, Jin YH. SARS-CoV-2 RNA dependent RNA polymerase inhibitors selected from a cell-based SARS-CoV-2 RNA dependent RNA polymerase activity assay system. Biomedicines. 2021;9(8):996-1010.
Crossref - Tian L, Qiang T, Liang C, et al. RNA-dependent RNA polymerase(RNA dependent RNA polymerase) inhibitors:the current landscape and repurposing for the COVID-19 pandemic. Eur J Med Chem. 2021;213:113201.
Crossref - Sahoo M, Jena L, Rath SN, Kumar S. Identification of suitable natural inhibitor against Influenza A(H1N1) Neuraminidase protein by molecular docking. Genomics Inform. 2016;14(3):96-103.
Crossref - Natesh J, Mondal P, Kaur B, Salam AAA, Kasilingam S, Meeran SM. Promising phytochemicals of traditional Himalayan medicinal plants against putative replication and transmission targets of SARS-CoV-2 by computational investigation. Comput Biol Med. 2021;133:104383.
Crossref - Maiti P, Nand M, Mathpal S, et al. Potent multi-target natural inhibitors against SARS-CoV-2 from medicinal plants of the Himalaya:a discovery from hybrid machine learning, chemoinformatics, and simulation assisted screening. J Biomol Struct Dyn. 2023;21:1-14.
Crossref - Melloul M, Chouati T, Hemlali M, et al. Genome sequences of the delta variant (B.1.617.2) and the kappa variant (B.1.617.1) detected in Morocco. Microbiol Resour Announc. 2021;10(39):e0072721.
Crossref - Altschul SF, Gish W, Miller W, et al. Basic local alignment search tool. J Mol Biol. 1990;215(3):403-410.
Crossref - Sievers F, Higgins DG. Clustal omega. Curr Protoc Bioinformatics. 2014;48(1):1-16.
Crossref - Buchan DWA, Jones DT. The PSIPRED protein analysis workbench:20 years on. Nucleic Acids Res. 2019;47(W1):W402-W407.
Crossref - Jones DT. Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol. 1999;292(2):195-202.
Crossref - Jones DT, Cozzetto D. DISOPRED3: precise disordered region predictions with annotated protein-binding activity. Bioinformatics. 2015;31(6):857-863.
Crossref - Nugent T, Ward S, Jones DT. The MEMPACK alpha-helical transmembrane protein structure prediction server. Bioinformatics. 2011;27(10):1438-1439.
Crossref - McGuffin LJ, Jones DT. Improvement of the GenTHREADER method for genomic fold recognition. Bioinformatics. 2003;19(7):874-881.
Crossref - Lobley A, Sadowski MI, Jones DT. pGenTHREADER and pDomTHREADER:new methods for improved protein fold recognition and superfamily discrimination. Bioinformatics. 2009;25(14):1761-1767.
Crossref - Bryson K, Cozzetto D, Jones DT. Computer-assisted protein domain boundary prediction using the Dom-Pred server. Curr Protein Pept Sci. 2007;8(2):181-188.
Crossref - Waterhouse A, Bertoni M, Bienert S, et al. SWISS-MODEL:homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46(W1):W296-W303.
Crossref - Bienert S, Waterhouse A, de Beer TAP, et al. The SWISS-MODEL repository-new features and functionality. Nucleic Acids Res. 2017;45(D1):D313D319.
Crossref - Studer G, Rempfer C, Waterhouse AM, et al. QMEANDisCo-distance constraints applied on model quality estimation. Bioinformatics. 2020;36(6):1765-1771.
Crossref - Biasini M, Bienert S, Waterhouse A, et al. SWISS-MODEL:modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014;42:W252-258.
Crossref - Benkert P, Biasini M, Schwede T, Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics. 2011;27(3):343-50.
Crossref - Kallberg M, Wang H, Wang S, et al. Template-based protein structure modeling using the RaptorX web server. Nat Protoc. 2012;7(8):1511-1522.
Crossref - McGuffin LJ, Atkins JD, Salehe BR, et al. IntFOLD:an integrated server for modelling protein structures and functions from amino acid sequences. Nucleic Acids Res. 2015;43(W1):W169-W173.
Crossref - Chen VB, Arendall WB, Headd JJ, et al. MolProbity:all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt1):12-21.
Crossref - Sigrist CJA, Cerutti L, Hulo N, et al. PROSITE: A documented database using patterns and profiles as motif descriptors. Brief Bioinform. 2002;3(3):265-274.
Crossref - Volkamer A, Kuhn D, Rippmann F, Rarey M. DoGSiteScorer:a web server for automatic binding site prediction, analysis and druggability assessment. Bioinformatics. 2012;28(15):2074-2075.
Crossref - Arcusa R, Villano D, Marhuenda J, CanoBego M, Cerda B, Zafrilla P. Potential role of ginger (Zingiber officinale Roscoe) in the prevention of neurodegenerative diseases. Front Nutr. 2022;9:809621.
Crossref - Kim S, Thiessen PA, Bolton EE, et al. PubChem substance and compound databases. Nucleic Acids Res. 2016;44(D1):D1202-D1213.
Crossref - Zhang MQ, Wilkinson B. Drug discovery beyond the ‘rule-of-five’. Curr Opin Biotechnol. 2007;18(6):478-488.
Crossref - Cheng F, Li W, Zhou Y, et al. AdmetSAR:a comprehensive source and free tool for assessment of chemical ADMET properties. J Chem Inf Model. 2012;52(11):3099-3105.
Crossref - Schneidman-Duhovny D, Inbar Y, Nussinov R, Wolfson HJ. PatchDock and SymmDock:servers for rigid and symmetric docking. Nucleic Acids Res. 2005;33(Suppl 2):W363-W367.
Crossref - Goddard TD, Huang CC, Meng EC, et al. UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Sci. 2018;27(1):14-25.
Crossref - Pettersen EF, Goddard TD, Huang CC, et al. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021;30(1):70-82.
Crossref - Trott O, Olson AJ. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comput Chem. 2010;31(2):455-461.
Crossref - Cossins EA, Sinha SK. The interconversion of glycine and serine by plant tissue extracts. Biochem J. 1996;101(2):542-549.
Crossref - Voet D, Voet JG. Three dimensional structures of proteins. In Recta P.(ed.) Biochemistry. 4th Ed. Wiley Press, USA. 2010:221-277.
- van Dijk E, Hoogeveen A, Abeln S. The hydrophobic temperature dependence of amino acids directly calculated from protein structures. PLoS Comput Biol. 2015;11:e1004277.
Crossref - Singh AK, Singh A, Singh R, Misra A. Remdesivir in COVID-19: A critical review of pharmacology, pre-clinical and clinical studies. Diabetes Metab Synd. 2020;14(4):641-648.
Crossref - Jahan R, Paul AK, Bondhon TA, et al. Zingiber officinale: Ayurvedic Uses of the Plant and In Silico Binding Studies of Selected Phytochemicals With Mpro of SARS-CoV-2. Nat Prod Commun. 2021;16(10):1-13.
Crossref
© The Author(s) 2024. Open Access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License which permits unrestricted use, sharing, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.