


Staphylococcus aureus is a major human pathogen that may cause a wide range of infections and is a frequent cause of soft tissue and bloodstream infections. It is a successful pathogen due to its collective virulence factors and its ability to evade the host immune systems. The review aims to highlight how S. aureus destroys and damage the host cells and explains how immune cells can respond to this pathogen. This review may also provide new insights that may be useful for developing new strategy for combating MRSA and its emerging clones such as community-associated methicillin-resistant S. aureus (CA-MRSA).
Staphylococcus aureus, pathogenesis, immune system, virulence, immune evasion
Initially described in 1878, staphylococci, Gram-positive cocci that have been implicated in infections involving multiple systems of the human body, including the skin and soft tissue, the skeletal system, the respiratory system, the bloodstream, and more recently, infections involving implanted medical devices1,2. Staphylococci are further classified as being coagulase-positive, primarily identifying Staphylococcus aureus (S. aureus), or coagulase-negative (CoNS)3. Staphylococcus epidermidis (S. epidermidis) is the most important and best-studied member of the CoNS group4.
S. aureus is facultative, non-spore-forming, and non-motile bacteria that have the ability to grow well on most cultural media. S. aureus is a common inhabitant of the human and animal body, particularly the nasal cavity and the skin4. Despite being a commensal in human and animals, S. aureus is a virulent bacterium that are able to produce number of toxins and enzymes which allow this bacterium to be able to invade human body and cause wide range of infections, some of which may be considered as life-threatening diseases (Table 1) in immunocompromised and immunocompetent individuals5, 6. Moreover, some strains of S. aureus are resistant to a wide range of antibacterial agents, some of which show multiple resistance (resistant to three or more classes of antibacterial agent), The most clinically important strains are the methicillin-resistant Staphylococcus aureus (MRSA) which are implicated in a large number of hospital and community acquired infections. The resistance towards antibacterial agents and at times multiple resistance make the treatment of these infections complicated and more challenging to achieve7. This review aims to provide and updated literature survey on epidemiology, virulence and pathogenicity as well as interaction mechanisms with host immune system by S. aureus.
Table (1):
Human infections caused by Staphylocccus aureus.
Infection site |
Clinical syndrome |
|---|---|
Skin and soft tissues |
Boils, abscesses, impetigo, wound infection, scalded skin syndrome, necrotizing fasciitis, cellulitis |
Bone |
Osteyomyelitis |
Joints |
Septic arthritis |
Blood |
Bacteremia, Toxic shock syndrome, septic thrombophlebitis |
Lung |
Pneumonia |
Brain |
Brain abscess, meningitis |
Heart |
Endocarditis |
Urinary tract |
Urinary tract infection |
Intestine |
Food poisoning |
Staphylococcus aureus Epidemiology
S. aureus infections may not be all reported, given the wide variety of community and hospital-acquired of infection, however, the number of reported cases is increasing worldwide, for instance, in the USA around 80461 cases of MRSA infections in 2011, resulting in a 11000 deaths6. Reported MRSA infections in Australia increased from 10.3% in 2000 to 16% in six years8. In Taiwan, dramatic increase of MRSA infection rose from 9.8% in 2000 to 56% in 2005, similar increases of S. aureus infections were also reported in Saudi Arabia, Lebanon, England, India6,8,9. It is important to mention that these figures represent over all infections, the epidemiological data of every type of Tong et al.5 has well documented S. aureus infections concluding that there is an increase of hospital-acquired infections particularly in the cases of infective endocarditis and infections related to prosthetic devices, moreover, an epidemic of community-associated skin and soft tissue infections controlled by certain virulence factors and resistance to antibiotics particularly β-lactam group5. Molecular epidemiology of S. aureus infections showed diversity of clones responsible for infections in every content, with the existence of predominant clones such as ST-80 which was found in North and South America and Europe. In Asia, high heterogenecity in terms of the clones associated with infection was highlighted. In general, it was suggested that low restrictions in the sales of antibiotics and/or prescribing antibiotics with proper laboratory analysis in some countries aided the increase of S. aureus infections as well the diversity of clones responsible for these infections8.
Staphylococcus aureus virulence factors
S. aureus infections occur when the mucosal or skin barriers are breached, following the successful invasion of the bacterium into the host tissues and its ability to escape the defensive barriers of the immune system to enter the bloodstream. S. aureus have an arsenal of virulence factors that play significant roles in the wide variety of infections and diseases in humans including animals. These factors can also support and provide protection for S. aureus to evade the host immune system recognition and their actions.
One of the most important virulence factors is surface proteins which promote and encourage the binding and the attachment processes of this bacterium to the host cells surfaces. To avoid recognition from the host immune cells, the surface proteins combined with the blood proteins that lead to aid the bacterium to survive and cause damage to host tissues10. Another vital factor is protein A, which is located in the cell wall of S. aureus which accurately anchored to the peptidoglycan pentaglycine bridges of the bacteria. It is also known as an IgG-binding protein which combines with the Fragment crystallisable (Fc) region of the antibody in order to cover the surface of the bacterium with IgG antibody to make the recognition of this organism by the immune cells difficult and hard to detect11,12.
The clumping factor A (ClfA) is another S. aureus virulence factor that is expressed by the surface of the bacteria cells. Also, it is known as fibrinogen binding protein that promotes the clotting process of blood cells and the damage process of tissues13. In addition, the polysaccharide capsule is considered as one of the most essential virulence factor which can contribute to assist the S. aureus surviving within the host cells by inhibiting the phagocytosis process by macrophage and dendritic cells10, moreover, S. aureus cells are capable of secreting several important toxins and enzymes such as; coagulase; DNAase; leukocidin; hemolysins; exfoliative toxin in order to promote bacterial penetration and help the bacterium to evade in to the host tissues (Table 2)10.
Table (2):
Staphylococcus aureus virulence factors.
Factor |
Effect / function |
|---|---|
Cell surface proteins |
Helping attachment to host tissues |
Polysaccharide microcapsule |
Resist phagocytosis |
Protein A |
Limits the host immune response |
Panton-Valentine leukocidin (PVL) |
Forming porins in cell membrane of host cells, resulting in cell death by leakage ot its contents |
Alpha hemolysin |
Red blood cells lysis |
Chemotaxis inhibitory proteins |
Inhibit the chemotaxis of neutrophil and monocytes |
Extracelleular adherence protein |
Play a role in adherence and invasion of host tissues and also has immune-modularity activity |
Proteases, lipases, nucleases, hyaluronatelyase, phspholipase C, elastase and Staphylokinase |
These enzymes contribute to host tissue destruction that facilitate the invation and penetration of the bacterium into host tissues |
Toxic shock syndrome toxin-1 (TSST-1) |
Causes toxic shock syndrome particularly in menstrual women |
Exfoliative toxin |
Causes scalded skin syndrome particularly affecting infants |
Enterotoxins |
Several types pf enetrotoxins (SEA, SEB, SEC, SED, SEG, SEH & SEI) causes food poisoning |
Clumping factor |
Clots blood plasma |
Adapted from [10, 11, 12, 13]
Moreover, S. aurues can generate another harmful type of toxins such as Panton-Valentine leukocidin (PVL) which cause pneumonia in children and Toxic Shock Syndrome Toxin-1 (TSST-1) which is associated with some cases of septicaemia due to the use of particular types of tampons (Table 2)13,10. A number of S. aureus strains have the ability to produce a pigment that known as staphyloxanthin which acts as an important virulence factor. This strain has an antioxidant role against reactive oxygen species used by the host immune cells, in order to help the bacterium to escape from killing action of immune cells14. Furthermore, S. aureus are capable of forming biofilms of various surfaces which associated generally with most indwelling medical devices problems such as heart valves and knee replacements and via this biofilm community resistance can be acquired to antibacterial agents through horizontal gene transfer12.
How Does S. aureus subvert immune responses?
When S. aureus invade the host tissues, the innate immune system responds rapidly as an early defense against the bacterial invasion. This system consists of three important parts; (i) the complement system, (ii) phagocytes and (iii) antimicrobial peptides. It also play many significant roles against the pathogen for instance; it summons the immune cells to the infection sites by producing chemical signaling molecules called cytokines, activates the complement system to recognize the microbe to encourage the clearance process of dead cells, and it activates the second line of the host defense which is known as the adaptive (specific) immune system through antigen presentation process, in order to help the body to eliminate the microbe and to generate long lasting immunity against it to prevent any challenge in the future by the same organism15,16.
The Complement System
The complement system is a group of proteins and proteolytic molecules that are found and circulate in the blood (Table 3). With S. aureus, complement employs some significant molecules to: (1) mark the microbe with C3b and iC3b to make its parts ready for phagocytosis through important immune cells such as; neutrophil and macrophage, and also to (2) seize the attention of phagocytes by small chemoattractant molecules such as: C3a and C5a which are produced during the activation of complement to facilitate phagocytosis as well15,17.
Table (3):
Functional protein classes in the complement system.
Types of protein classes |
Examples |
|---|---|
Binding to antigen: antibody complexes and pathogen surfaces |
C1q |
Binding to mannose on bacteria |
MBL |
Activating enzymes |
C1r, C1s, C2b, Bb, D, MASP-1 and MASP-2 |
Membrane-binding proteins and opsonins |
C4b and C3b |
Peptide mediators of inflammation |
C5a, C3a and C4a |
Membrane-attack proteins |
C5b, C6, C7, C8 and C9 |
Complement receptors |
CR1, CR2, CR3, CR4 and C1qR |
Complement-regulatory proteins |
C1INH, C4bp, CR1 and MCP |
Adapted from [25]
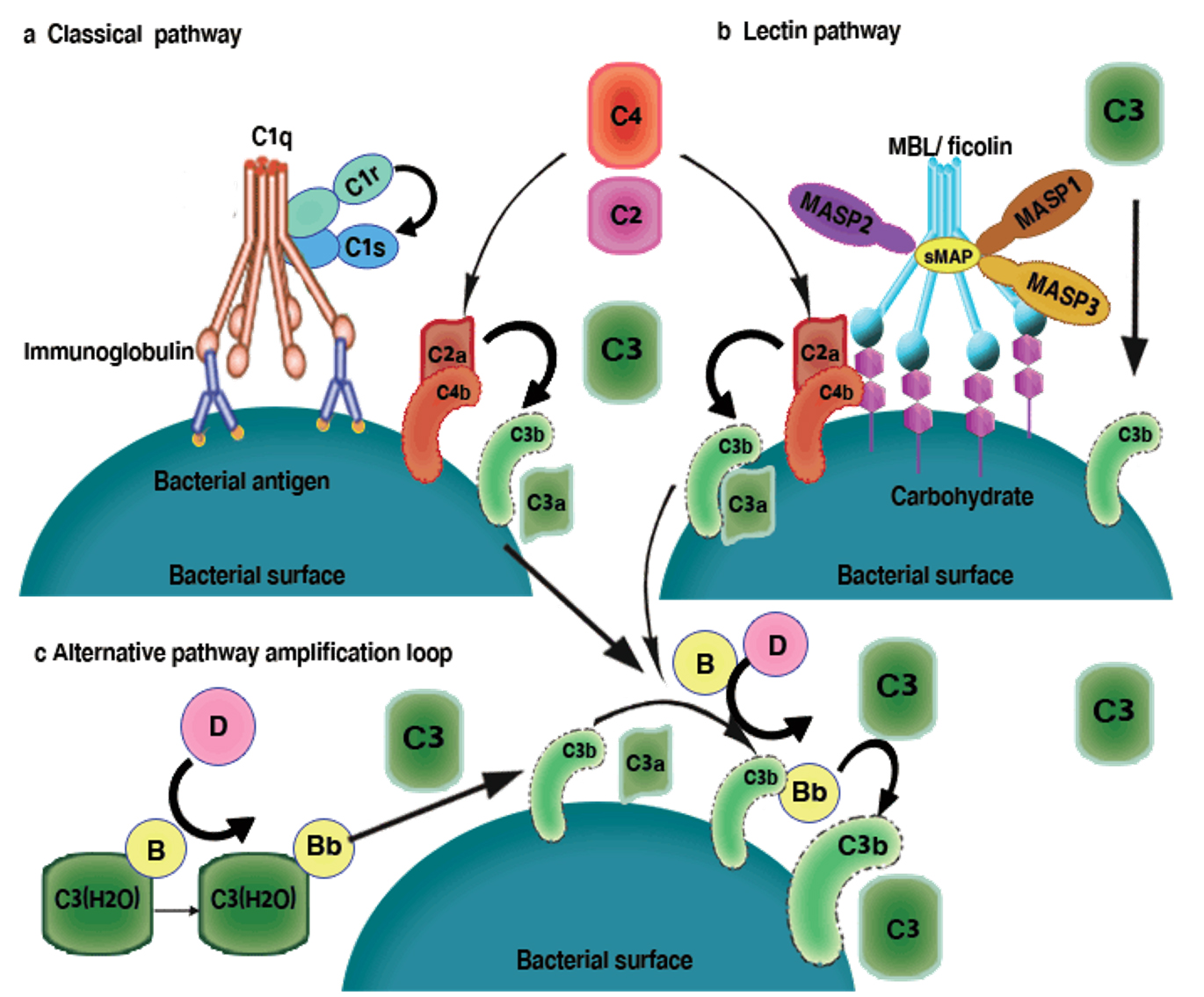
Fig. 1. Complement activation pathway: (A) The classical pathway is initiated by the binding of the C1 complex to antibodies that are bound to antigens on the surface of bacteria. The C1 complex consists of C1q and tow molecules each of C1r and C1s. The binding of the recognition subcomponent C1q to the Fc portion of immunoglobulins results in autoactivation of the serine protease C1r. Then C1r cleaves and activate C1s. which translates the activation of the C1 complex into complement activation through the cleavage of C4 and C2 to form C4bC2a enzyme complex. C4bC2a acts as a C3 convertase and cleave C3 which results in products that bind to, and cause the destruction of, invading bacteria. (B) The lectin pathway is initiated by the binding of either mannose binding lectin (MBL) or ficolin -associated with MBL-associated serine protease1 (MASP2), MASP2, MASP3 and small MBL-associated protein (sMAP) – to an array of carbohydrate groups on the surface of a bacteria cell. Similar to C1s, MASP2 is responsible for the activation of C4 and C2, which leads to the generation of the same C3 convertase (C4bC2a). As in the classical pathway, C3 convertase cleaves C3 to C3b and the chemo attractant peptide C3a. The C3b-C2a-C4b complex then cleaves C5 to C5a and the chemo attractant peptide C5b, which stimulates assembly of factors C6, C7, C8 and C9. MASP1 is able to cleave C3 directly. (C) The alternative pathway is initiated by the low grade activation of the C3 by hydrolysed C3 (C3(H2O)) and activated factor B (Bb). The activated C3b binds factor B (B), which is then cleaved into Bb by factor D (D) to form the alternative pathway C3 convertase, C3bBb. Once C3b is attached to the cell surface, the amplification loop consisting of the alternative pathway components is activated, and the C3 convertase enzyme cleaves many molecules of C3 to C3b, which bind covalently around the site of complement activation [15, 17,48, 49].
This system is initiated and activated by three significant pathways: (a) the classical, (b) the alternative and (c) and the lectin pathways (Fig. 1). All the three pathways come together at the formation of a surface bound biomolecular enzyme known as C3 convertases. This enzyme stimulates and enhances the activation of complement system by cleaving the complement protein C3 to form C3a and C3b which are required to promote and facilitate any additional essential activation events such as; opsonization and phagocytosis15-17.
S. aureus Complement avoidance mechanisms
It has been shown that S. aureus developed different mechanisms to avoid the action of the complement system by producing a number of proteins that can change and affect the stages of the complement cascade, these mechanisms include; preventing complement identification; cleavage of complement proteins; and/or inhibit the interaction of complement receptors on phagocytes18,19.
Although, the classical pathway C1 complex has the ability to recognize the microbe bound IgG and IgM antibodies and the lectin pathway (mannose-binding lectins and ficolins) bind the saccharide elements of microbe, S. aureus produce two surface proteins that can damage and harm the IgG function (Fig 2.a), the first protein is the Staphylococcal protein A (SpA) and the second protein is known as the Staphylococcal immunoglobulin binding protein (Sbi). Staphyloccocal protein (SpA) is a protein located on the surface of the bacterium that consists of four or five immunoglobulin binding domains. Every domain is able to combine with the Fc parts of IgG antibody, thus inhibiting the interaction with Fc receptors on neutrophils in vitro17-19.
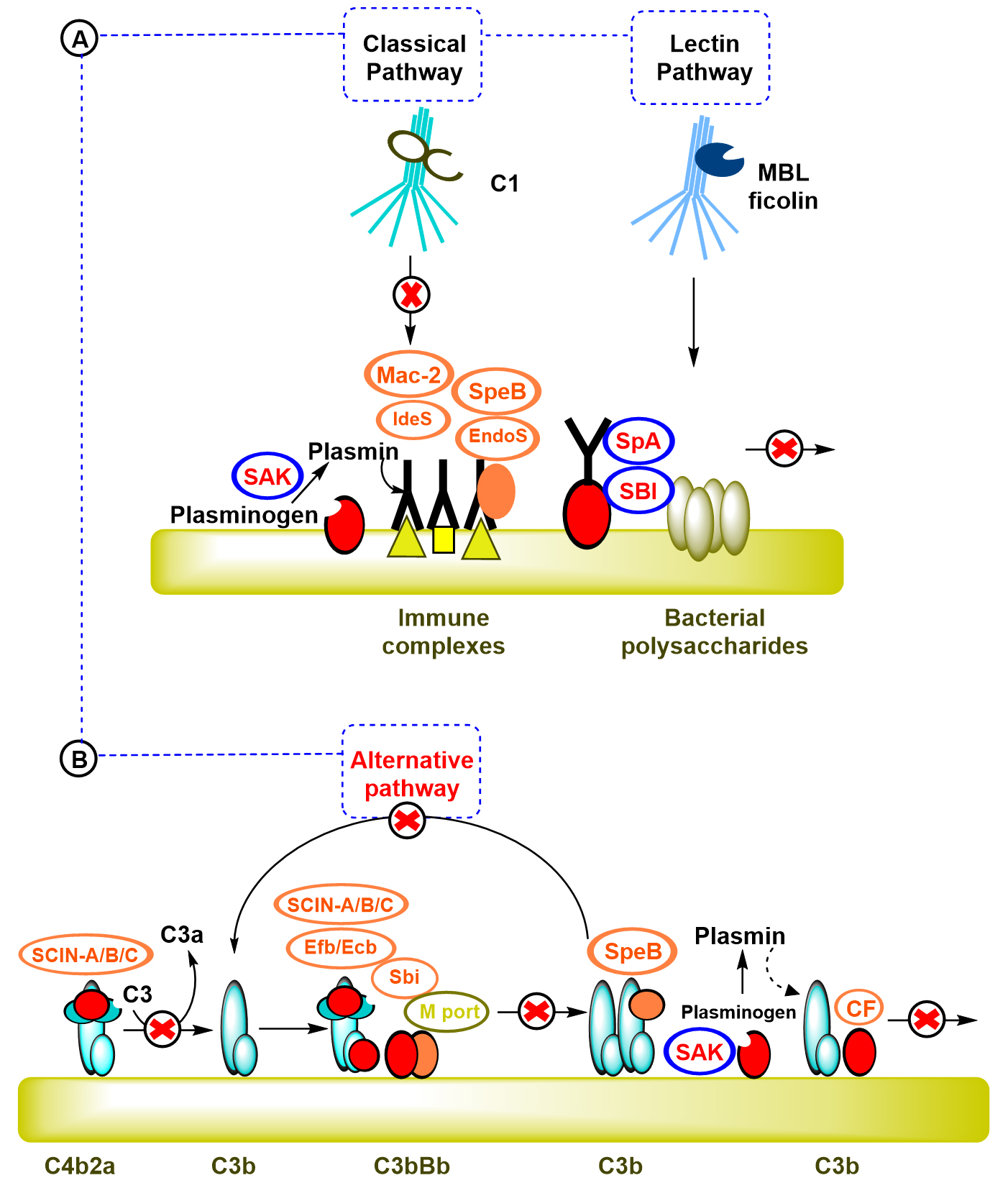
Fig. 2. Staphylococcus evasion of complement recpgnition: (A)- S. aureus produce two surface proteins that can damage and harm IgG function; Staphylococcal protein A (SpA) and Staphylococcal immunoglobulin binding protein (Sbi). (B)- Presence of the C3 convertases is significant for the activation of complement system and for the response of host immune cells. S. aureus use three ways to affect and change this vital step in the complement cascade [17, 18, 19].
Staphylococcal Sbi is made of four parts of Sbi-I and Sbi-II which are also able to bind with IgG. Besides blocking Fc-receptor-mediated phagocytosis, Sbi has been shown to play some roles in blocking the binding of C1q and subsequent activation of the complement classical pathway18. Furthermore, S. aureus uses another approach to evade the recognition of the complement system by removing and degrading the opsonic molecules (the molecules that are able to bind with both antigen and receptors of phagocytic cells like C3b component of complement system) from it surface through proteolytic process. In addition, S. aureus produce a staphylokinase (SAK) which is an anti-opsonic protein, in order to stimulate and encourage surface bound plasminogen into plasmin, which make the bacteria able to invade and infect the host tissues17. In fact, the presence of the C3 convertases is significant for the activation of complement system and for the response of host immune cells. S. aureus uses three ways to affect and change this vital step in the complement cascade (Fig. 2.b)17,18.
The Cleavage of C3 convertase
The S. aureus clumping factor A (ClfA) is a surface protein which can join the human C3b protease factor I (fI), thereby enhancing cleavage of surface-bound C3b into iC3b in vitro20.
Direct inactivation of C3 convertases
Convertases are the major complement target among S.aureus immune evasion strategies. S. aureus produces five unlike molecules which work directly to prevent the action of these important enzyme mixtures. Staphylococcal complement inhibitor (SCIN) and its homologues SCIN-B and SCIN-C are extremely efficient C3 convertase inhibitors which inhibit the alteration of C3, later phagocytosis and C5a formation in vitro at low concentrations18.
Besides that, the alternative pathway C3 convertase made of a vital cofactor called C3b which connected to the protease subunit (Bb) loosely18. The action of SCIN on the classical pathway convertase remains to be resolved but seems to be caused by a stabilizing mechanism as well21. In addition, the alternative pathway convertase can be changed via the extracellular fibrinogen-binding protein (Efb) and the extracellular complement-binding protein (Ecb) by binding the convertase to the C3b particle directly20. The crystal structures of both molecules in complex with the C3d domain of C3 have revealed their exact binding sites21.
Modulating human convertase regulators
In order to protect from unnecessary action of the complement system, humans produce complement regulators which reduce and decrease the activity of the convertase22. On the other hand, a large number of microbes generate molecules which interfere with the function of theses regulators. In addition to its two IgG binding fragments, Sbi-III and IV that can also bind to the C3, the staphylococcal IgG-binding molecule Sbi plays several important roles in the alteration of the complement23. Moreover, Sbi has the ability to bind the human complement regulators such as; factor H (FH) and factor H-related proteins in order to make a constant tripartite compound with FH and C3. In general, these actions lead to inhibit the activity of the alternative pathway in vitro24.
Phagocytosis
Phagocytosis is a common mechanism that involved in the immune response to eliminate pathogens or foreign particle may be a toxin invading the body. It is activated through the binding to pathogen-associated molecular patterns (PAMPS), which leads to the activation of nuclear factor kappa B (NF-κB) that is important to control and regulate the response of the immune system25. Furthermore, there are types of the immune cells that have the ability to engulf microbial pathogens in order to remove them from the host, these cells include; neutrophils, macrophages and dendritic cells26. Moreover, complement system plays an important role in phagocytosis by facilitating the uptake of the pathogen by phagocyte cells. This process occurs through specific complement receptors (CRs) such as; CR1 and CR2 (Table 4), these receptors bind the microbe to make it ready for phagocytosis25,26.
Table (4):
Types and functions of complement receptors.
Receptor |
Specificity |
Function |
Target cells |
|---|---|---|---|
CR1 (CD35) |
C3b, C4b ,iC3b |
Promotes C3b and C4b decay Stimulates phagocytosis Erythrocyte transport of immune complexes |
Erythrocyte, macrophage, monocytes, leukocytes, B cells |
CR2 (CD21) |
iC3b |
Part of B-cell co-receptor |
B cells |
CR3 (Mac-1) (CD11b/CD18) |
iC3b |
Stimulates phagocytosis |
Macrophages, monocytes, leukocytes |
CR4 (gp150,95) (CD11c/CD18) |
iC3b |
Stimulates phagocytosis |
Macrophages, monocytes, leukocytes, dendritic cells |
C5a receptor |
C5a |
Binding of C5a activates G protein |
Endothelial cells, mast cells, phagocytes |
C3a receptor |
C3a |
Binding of C3a activates G protein |
Endothelial cells, mast cells, phagocytes |
Adapted from [25]
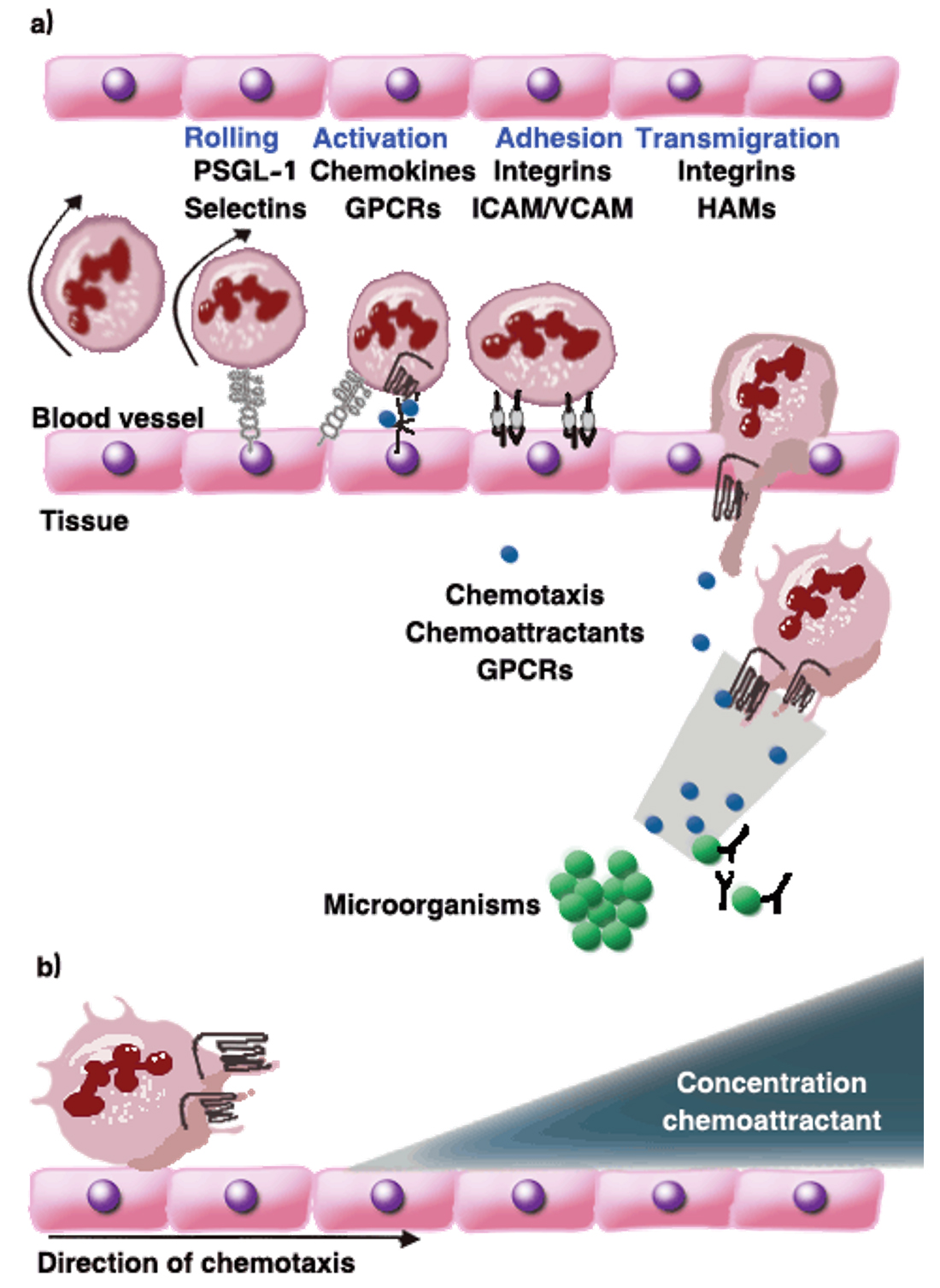
Fig. 3. Phagocyte extravasation: (a) for phagocytes to reach infection sites, they need to extravasate from the blood vessels into the tissue. The successful recruitment of circulating immune cells depends on the productive interaction of leukocytes with the endothelial cells lining the vessel wall and it is a multistep process. Phagocytes tether and roll on activated endothelium through transient interactions of PSGL-1 and selectins. Following stimulation of the cells by endothelium bound chemokines, integrins are activated that mediate the firm cellular adhesion to intercellular adhesion molecule-1 (ICAM-1) and vascular adhesion molecule-1 (VCAM-1) presented by the vessel wall. The phagocytes then transmigrate through the endothelium into the underlying tissue .The cells are directed to the site of infection through the sensing of chemoattractant gradients by GPCRs. Here, chemokines and bacteria and complement derived products are important. The pathogen is cleared by phagocytosis upon recognition of the bacterium opsonized by antibodies and/or complement components. (b) At the infection site, infected cells and the surrounding tissues release chemoattractants. These secreted chemoattractants form a concentration gradient and attract leukocytes, which move through the gradient towards the higher concentrations, a process called chemotaxis. Leukocytes move through the gradient toward the higher concentrations, a process called chemotaxis [25, 26, 17].
In addition, at the early stage of the immune response to the site of infection, the pathogen and its fragments are engulfed by antigen-presenting cells and other immune cells such as macrophages and neutrophils and transferred to the lymph nodes. This leads to the activation of the B cells to discriminate and to produce antibodies in order to reduce the effects of the bacterial toxins and to encourage and promote the phagocytosis of the pathogen26. Furthermore, following the entry of S. aureus to the host tissue, the response of neutrophils and macrophages initiate which is critical and essential to promote and assist the body in removing and eliminating the pathogens during the phagocytosis21. (Fig. 3.a).
These cells are called via an important chemoattractants such as C5a; leukotriene B4; and chemokines such as CXCL8 which also known as interleukin-8 (IL-8) in order to direct their actions to the site of infection27. This chemoattractants formed through activated host cells and some of them produced as an activated component of the complement system (C5a) at the time of identification of a preserved structure present on the bacterium28. All these chemoattractants trigger phagocytes by attaching to membrane bound receptors called G protein-coupled receptors (GPCRs). Both chemoattractants and GPCRs have an essential role which is directing the innate defence cells against the invading microbe (Fig. 3.b). As a result S. aureus has a wide range of strategies to evade and avoid phagocytic activity28,29.
S. aureus phagocytosis avoidance mechanisms
Inhibition of neutrophil chemotaxis
A large number of S. aureus strains (about 60%) secrete a chemotaxis inhibitory protein (CHIPS) which can combine with the C5a receptor (C5aR) and the formyl-peptide receptor (FPR) in order to block the binding of any related agonist (Fig. 4), the C5aR- and FPR-binding activities of CHIPS were separated by specific amino-acid substitutions and the specificity of blocking monoclonal antibodies19,30. The intercellular adhesion molecule-1 (ICAM-1) on the endothelial cell surface is one of the several ligands recognized by the extracellular adherence protein Eap (or else called the main histocompatibility class II analogue protein Map)30.
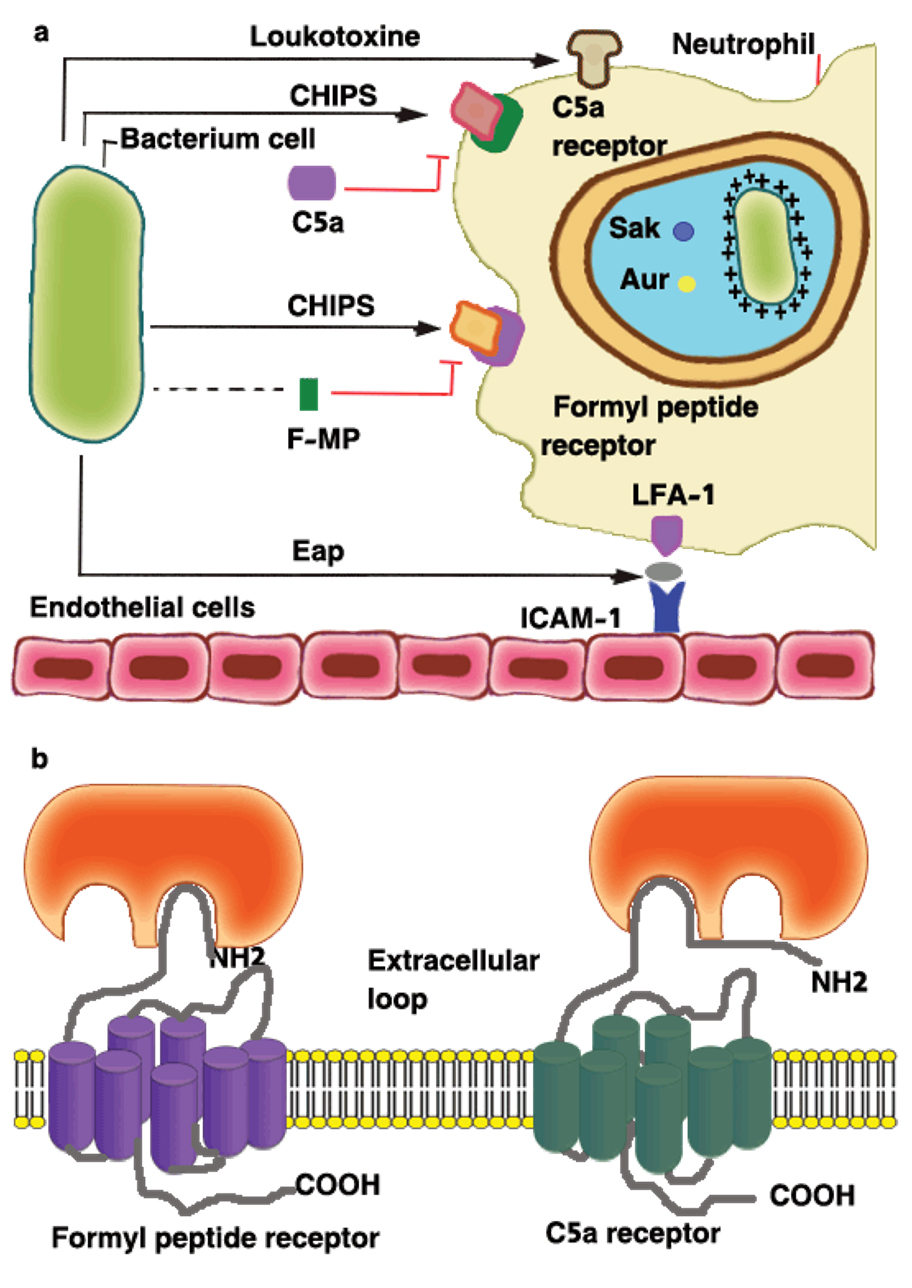
Fig. 4. Inhibition of the neutrophil response to infection: (a) the chemotaxis inhibitory protein of staphylococci (CHIPS) and the extracellular adherence protein (Eap) interfere with neutrophil chemotaxis and extravasation. Resistance to killing by antimicrobial peptides in the neutrophil phagosome is promoted by D-alanine and L-lysine modifications to cell-wall components (indicated by +), by secretion of staphylokinase (Sak) and aureolysin (Aur), and by the creation of ‘spacious’ phagosomes in which bacteria can survive. The pore-forming leukotoxins are shown by the mushroom-shaped insertion in the neutrophil membrane. (b) Model for interactions between CHIPS and the formyl peptide receptor (FPR) and C5a receptor. Two distinct but closely linked binding domains in CHIPS are indicated, one for the extreme N terminus of FPR involving residues F1 and F3, the second for a domain located between residues10–20 of the C5a receptor. FMP, N formyl-methionyl peptide; ICAM-1, intercellular adhesion molecule-1; LFA-1, lymphocyte function associated antigen [19, 30, 48].
Resistance to phagocytosis
S. aureus produces many significant factors to avoid and evade from phagocytosis. It expresses anti-opsonic proteins bound to the surface and a capsule of polysaccharides which: (a) interfere with antibodies and with the formation of complement through classical and alternative pathways, (b) inhibit their interaction to neutrophil complement receptor and Fc receptor. Therefore, efficient phagocytosis by neutrophils that requires recognition of bound complement proteins and antibody is compromised25. Moreover, S. aureus has some mechanisms which help and assist the organism to evade the killing of phagocytic cells, including interference with endosome fusion and release of antimicrobial substances by factors that are dependent on the global regulator SarA31.
Protein A
Is a surface protein that consists of four or five domains which can bind to the Fc region of IgG antibody. Protein A interact with IgG in order to cover the cell surface with IgG molecules which are in the incorrect orientation easy to recognize by the Fc receptor of neutrophil (Fig. 5).This action can clarify the anti phagocytic role of protein A and its effects on S. aureus infections32,33.
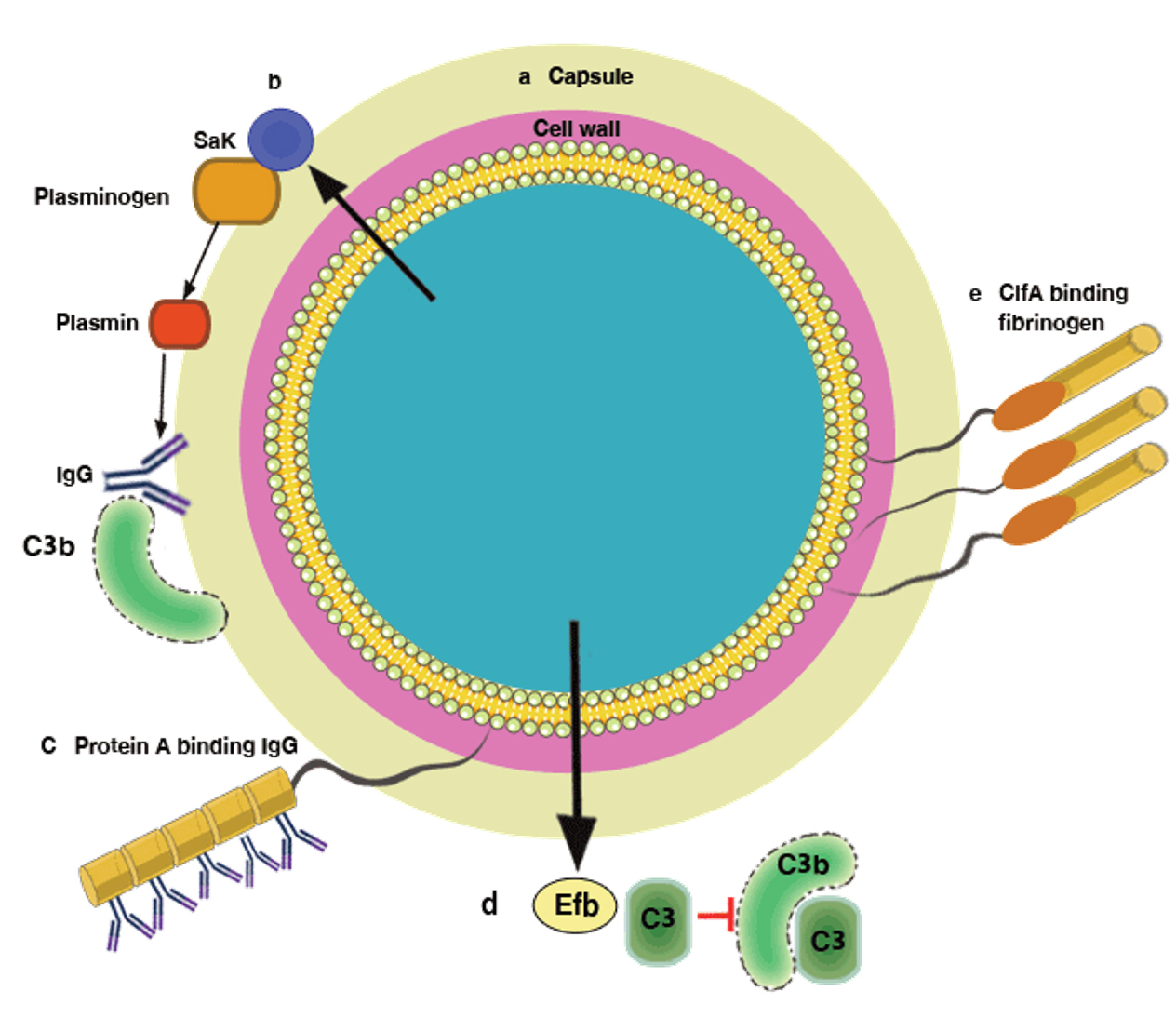
Fig. 5. Mechanisms by which Staphylococcus aureus avoids opsonophagocytosis: (a) the capsular polysaccharide, which can compromise neutrophil access to bound complement and antibody; (b) the extracellular staphylokinase (Sak), which activates cell-bound plasminogen and cleaves IgG and C3b; (c) protein A with 5 IgG Fc-binding domains; (d) fibrinogen-binding protein (Efb), which binds complement factor C3 and blocks its deposition on the bacterial cell surface. Complement activation beyond C3b attachment is prevented, thereby inhibiting opsonization. (e) ClfA, which binds the γ chain of fibrinogen [19, 21, 30, 33, 34].
Clumping factor A (ClfA)
Is the dominant fibrinogen-binding protein present on the surface of S. aureus cells in the stationary phase of growth. In the murine model the ClfA is a virulence factor for sepsis and arthritis34. Virulence was thought to increase during the bacteremic stage of the infection as well as during the growth of infected joints, since bacterial cells were coated with fibrinogen (Fig. 5), which in turn inhibited deposition of, or accessibility to opsonins30,34. This notion is supported by the observation that ClfA protects S. aureus from phagocytosis by murine macrophages46 and by human neutrophils and that defense is at least partly dependent on fibrinogen21,34. Furthermore, ClfB and fibronectin-binding proteins can also bind fibrinogen, to shield the bacterium through the exponential phase of growth, when the expression of such proteins is larger than ClfA34.
Capsule
Is an important factor which expresses by S. aureus to inhibit the process of phagocytosis. The expression of type 5 and type 8 capsules is associated with increase virulence in animal infection models35. It has been considered that the presence of the capsule lead to reduce the ability of neutrophils to uptake the pathogen cells in the presence of normal serum opsonins (Fig. 5), meaning that capsule act as anti-opsonic19.
Toxins that kill leukocytes
S. aureus is a common pathogen that has the ability to secrete and produce toxins which contribute to the damage of host cells membranes. The expression of cytolytic toxins that damage leukocytes contributes to development of abscesses by the killing of neutrophils that are attempting to engulf and kill the bacteria36. Cytolytic toxins forming β-barrel pores in target cell cytoplasmic membranes cause leakage, and eventually lysis. The representative of this class is the α-toxin, which is secreted as a monomer paired with a heptamer in the membrane, with β-strands assembled in a 14-stranded β-barrel pore from each monomer36,37.
The two-component leukotoxins consist of two subunits, which are separately secreted and configured into hexameric or heptameric oligomers with a great leukocyte affinity. There are 4 different types of bicomponent leukotoxin, y-toxin (Hlg), Panton–Valentine leukocidin (PVL), leukocidin E / D, and leukocidin close to M / F-PV. The γ-toxin lyses both erythrocytes and leukocytes, whereas PVL is toxic only for leukocytes36,38.
Other evasion mechanism of S. aureus
Immunomodulatory molecules
Protein A
In addition to its immunoglobulin binding ability, Protein A is an important immunomodulatory molecule owing to its capacity to combine with the VH3 region that is next to the antigen-binding domain of IgM molecules located on the B lymphocyte surface (Fig. 6). Those cells carrying VH3 IgM are induced to proliferate and undergo apoptosis, resulting in depletion of a large proportion of potential antibody-secreting B cells in the spleen and bone marrow39,40.
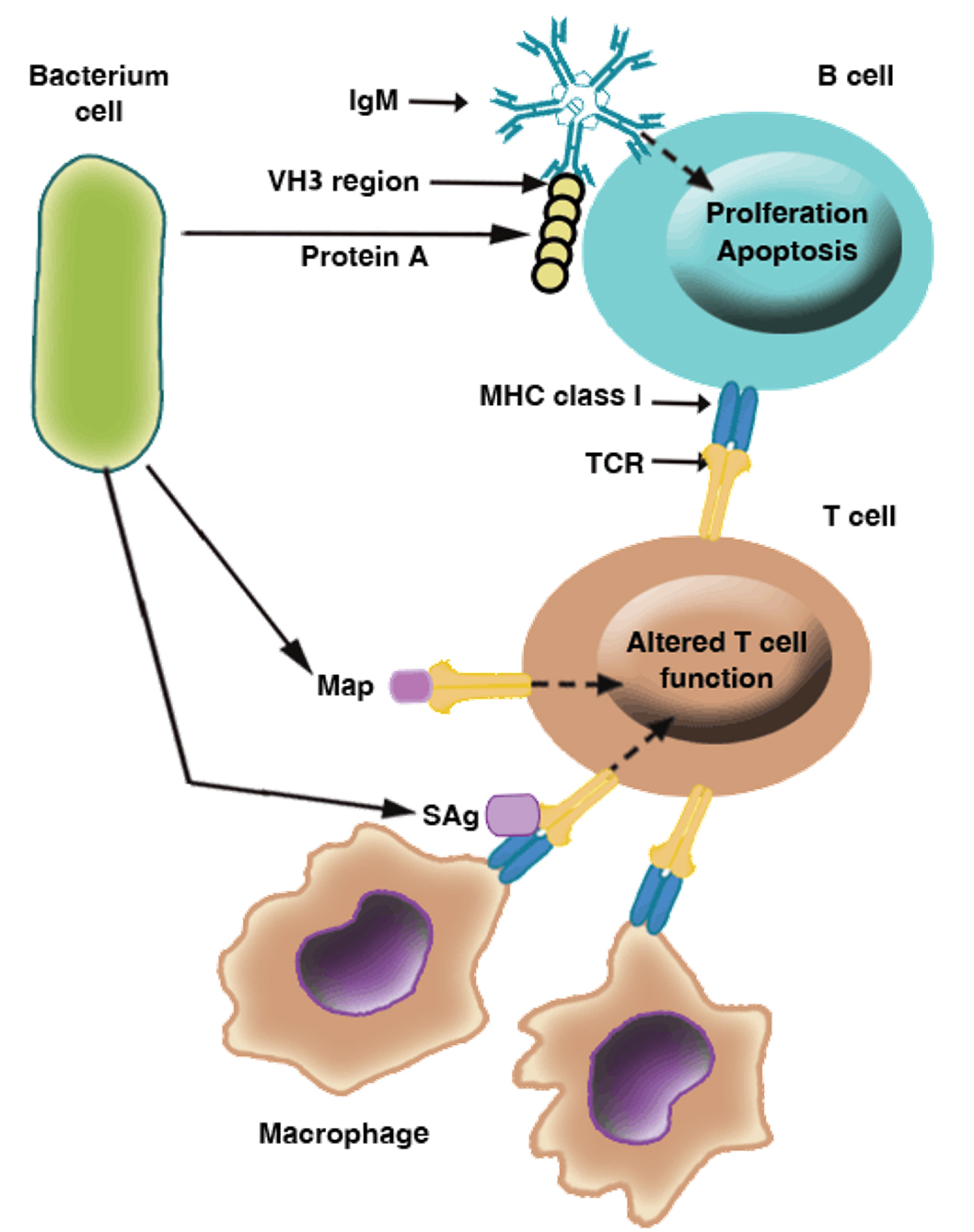
Fig. 6. Mechanisms of immunosuppression mediated by Staphylococcus aureus: This figure illustrates examples of immunomodulatory molecules used by S. aureus to alter the host immune response, including the superantigens (sAgs) enterotoxins and toxic shock syndrome toxin-1 that bind the MHC class II receptor to T-cell receptors; protein A, which binds immunoglobulin M (IgM) VH3 on B cells; and the MHC class II analogue protein Map, which binds the T-cell receptor (TCR) [36, 38, 39, 41].
Enterotoxins and TSST-1
S. aureus produces toxins which work as superantigens such as toxic shock syndrome toxin-1(TSST-1), the expression of these antigens in the host cell prevents the development of the immune response such as activation of B and T cells or formation of antibodies (Fig. 6)36,38. Furthermore, Antigen-specific T cells are unable to reproduce in response to antigens that are normally presented by MHC class II due to a phenomenon called anergy41. Therefore, immune- suppression occurred due to the failure of the antibody response. This seems to be important to prevent the production of antibodies to superantigenic toxins themselves36,38,41.
Secretable expanded repertoire adhesive molecules (SERAM)
S. aureus generates some bacterial proteins that anchored in the cell wall which has a significant role in mediating bacterial adherence to host cells and to the components of the extracellular matrix (ECM). The general roles of SERAM (Fig. 7) can be described as: (1) to facilitate bacterial adhesion to host molecules, cells, or tissues, (2) to interact with a broad array of host ligands, thereby sharing diverse activities in that they typically interfere with host defense mechanisms42,43.
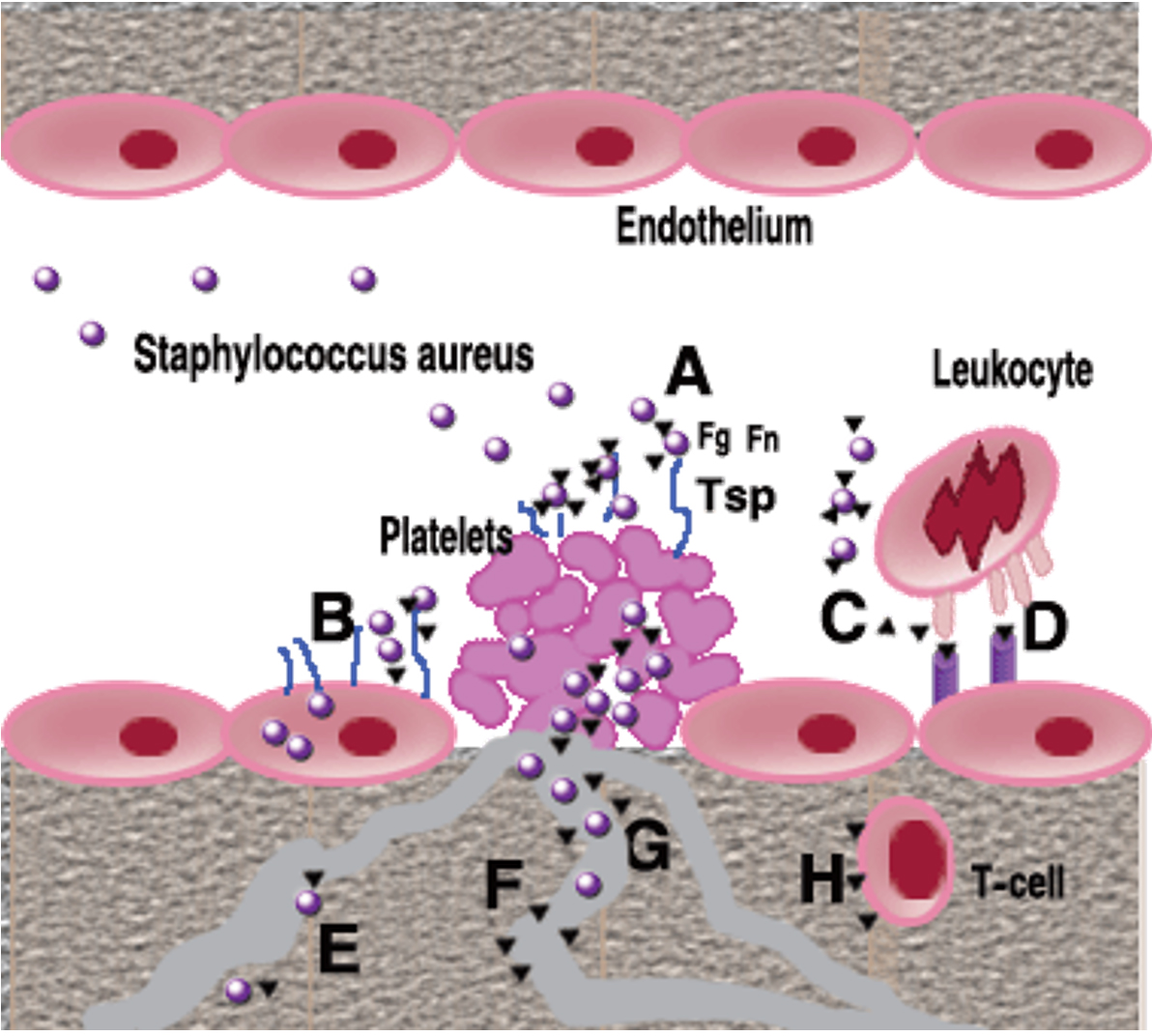
Fig. 7. Role of SERAM in endovascular Staphylococcus aureus infection: During wound infection, S. aureus organisms secreting and/or decorated by secretable extended repertoire adhesion molecules SERAM) (black triangles) gain access not only to the intact vessel wall, but also to subendothelial matrix and activated platelets and will form a bacteria-entrapped platelet clot (A). SERAM bind to various extracellular matrix components including fibrin (ogen) (Fg), fibronectin (Fn) or thrombospondin (Tsp) that are present in this vegetation and thereby augment bacterial attachment through molecular bridging mechanisms (Α). In addition, SERAM may facilitate bacterial binding to Fn expressed on endothelial cells resulting in enhanced uptake of microorganisms (Β). Through interaction with complement factor C3, the extracellular fibrinogen binding molecule (Efb) may interfere with opsono-phagocytosis (C). Among other interactions with host factors, the extracellular adhesion protein (Eap) binds to ICAM-1 and other ligands on endothelial cells thereby inhibiting leukocyte adhesion and preventing their extravasation (D). After passaging through the endothelial cell layer and/ or the extracellular matrix stroma, S. aureus may gain access to other capillaries, arterioles and venules, attach to the vessel wall and start disseminating from focal sites of infection (E) Eap may inhibit vascular cell proliferation and angiogenesis possibly via direct interference with agonist-stimulated endothelial functions (F), while Efb interacting both, with platelets and with fibrinogen, may interfere with fibrin formation resulting in altered wound healing (G). Finally, Eap interferes with T-cells function shifting towards a Th2-cell response and reducing delayed- type hypersensitivity reactions [42, 43].
Other proteins involved in immune-evasions mechanisms
Furthermore, some studies has shown new functions of some bacterial proteins such as extracellular matrix binding protein (Emp) and extracellular adherence protein (Eap) which play significant roles in S. aureus pathogenesis due to its ability to bind the host components or block some steps in wound healing process. Eap also known as a MHC class II analogous protein. It has a very wide spectrum of binding interactions to components of the host. However, some strains of S. aureus loss the ability to express Eap therefore these strains cannot colonize or invade host tissues44,45. This protein can also combine with endothelial ICAM-1 in order to inhibit the intraction between the ICAM-1 and its integrins Mac-1 and LFA-1. Thus, Eap completely destroyed the adhesion systems of respective functional leukocyte, i.e. strong adhesion and endothelial transmigration42. In addition to Eap anti-adhesive and anti-migratory function, it has been considered that Eap has an anti-inflammatory role by inhibiting neutrophils and T cells to get to the site of infections to prevent their effects against the bacterium. It also has been described that Eap has an immunomodulatory activity by increasing the synthesis of interleukin (IL)-4 syntheses which is vital for the differentiation of T-cells into Th2-cells in the response of the immune cells in order to down regulate and reduce the response of T-cells to facilitate the intracellular survival of S. aureus46-49.
It seems clear that S. aureus is a widespread organism that lives as a normal flora in the nose and on the skin of humans and animals. It is a very successful pathogen because of its ability to express and produce virulence factors to evade the recognition and the killing of the host immune cells. The bacterium infection starts through its entry to the host tissue which leads to stimulate and induce the innate immune response. The activation of this system occurred via specific pathways which lead to activate other important cells such as neutrophils and macrophages in order to eliminate and remove the pathogen from the host. The function of these cells initiated by the complement system which plays a significant role by producing chemoattarctant (chemotactic fators, chemokines, and complement factors etc.) in order to label and opsonise the pathogen for phagocytosis.
However, it has been shown that S. aureus improved many ways to avoid the effects of phagocyte cells by producing some proteins such as SpA and Sbi to inhibit and block the recognition of complement and their receptors interaction on phagocytes. Also, S.aureus can inhibit neutrophil chemotaxis by producing CHIPS that can interfere with some important receptors such as C5a receptor protein to prevent the binding of any related agonist. This pathogen can also produce super antigens toxins to make the response of the immune system difficult to achieve and hard to occur. Furthermore, it has been described that S. aureus can kill the leukocytes directly by secreting important cytolytic toxins such as PVL and leukocidin E/D. Moreover, SERAM, Eap and Emp are ubiquitous proteins that can help and assist the bacteria to survive and cause infections.
ACKNOWLEDGMENTS
None.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
AUTHORS’ CONTRIBUTION
All authors have made substantial, direct, and intellectual contribution to the work and approve it for publication.
FUNDING
None.
ETHICS STATEMENT
This article does not contain any studies with human participants, or animals performed by any of the authors.
AVAILABILITY OF DATA
All datasets generated or analyzed during this study are included in this manuscript
- Lowy FD. Staphylococcus aureus infections. New Eng J Med. 1998;339(8):520-532.
Crossref - Pollitt EJG, Szkuta PT, Burns N, Foster SJ. Staphylococcus aureus infection dynamics. PLoS Pathog. 2018;14(6):e1007112.
Crossref - Kloos WE, Schleifer KH. Gram-positive cocci. In: Bergey’s Manual of Systematic Bacteriology. Sneath PHA, Mair NS, Sharpe ME. (Eds.). 1986:1015-1022, Willians & Wilkins. USA.
- Oliveira WF, Silva PM, Silva RC, et al. Staphylococcus aureus and Staphylococcus epidermidis infections on implants. J Hospital Infect. 2018;98(2):111-117.
Crossref - Tong SYC, Davis JS, Eichenberger E, Holland TL, Fowler VG. Staphylococcus aureus infections: epidemiology, pathophysiology, clinical manifestations and management. Clin Microbiol Rev. 2015;28(3):603-661.
Crossref - Li Z. A review of Staphylococcus aureus and the emergence of drug-resistant problem. Adv Microb Physiol. 2018;8:65-76.
Crossref - Abulreesh HH, Organji SR, Osman GE, et al. Prevalence of antibiotic resistance and virulence factors encoding genes in clinical Staphylococcus aureus isolates in Saudi Arabia. Clin Epidemiol Glob Health. 2017;5(4):196-202.
Crossref - Junie LM, Jeican II, Matros L, Pandrea SL. Molecular epidemiology of the community-associated methicillin-resistant Staphylococcus aureus clones: a synthetic review. Clujul Med. 2018;91(1):7-11.
Crossref - Moghnieh R, Alothman AF, Althaqafi AO, et al. Epidemiology and outcome of invasive fungal infections and methicillin-resistant Staphylococcus aureus (MRSA) pneumonia and complicated skin and soft tissue infections (cSSTI) in Lebanon and Saudi Arabia. J Infect Public Health. 2017;10(6):849-854.
Crossref - Gnanamani A, Hariharan P, Paul-Satyaseela M. Staphylococcus aureus: Overview of bacteriology, clinical diseases, epidemiology, antibiotic resistance and therapeutic approach. In: Frontiers in Staphylococcus aureus. Enany S. Alexandar LC. (Eds.). Intech Open. 2017:4-28.
Crossref - Bien J, Sokolova O, Bozko P. Characterization of virulence factors of Staphylococcus aureus: novel function of known virulence factors that are implicated in activation of airway epithelial proinflammatory response. J Pathog. 2011;2011:601905.
Crossref - Shettigar K, Murali TS. Virulence factors and clonal diversity of Staphylococcus aureus in colonization and wound infection with emphasis on diabetic foot infection. Eur J Clin Microbiol Infect Dis. 2020;39(12):2235-2246.
Crossref - Lacey KA, Geoghegan JA, McLoughlin RM. The role of Staphylococcus aureus virulence factors in skin infection and their potential as vaccine antigens. Pathog. 2016;5(1):22.
Crossref - Kane TL, Carothers KE, Lee SW. Virulence factor targeting of the bacterial pathogen Staphylococcus aureus for vaccine and therapeutics. Curr Drug Targets. 2018;19(2):111-127.
Crossref - Degn SE, Thiel S. Humoral pattern recognition and the complement system. Scand J Immunol. 2013;78(2):181-193.
Crossref - Heesterbeek DAC, Angelier ML., Harrison RA, Rooijakkers SHN. Complement and bacterial infections: from molecular mechanisms to therapeutic applications. J Innate Immun. 2018;10(5-6):455-464.
Crossref - de Jong NWM, van Kessel KPM, van Strijp JAG. Immune evasion by Staphylococcus aureus. Microbiol Spectr. 2019;7:618-639.
Crossref - Laarman A, Milder F, van Strijp JAG, Rooijakkers S. Complement inhibition by gram-positive pathogens: molecular mechanisms and therapeutic implications. J Molec Med. 2010;88(2):115-120.
Crossref - Guerra FE, Borgogna TR, Patel DM, Sward EW, Voyich JM. Epic immune battles of history: neutrophils vs. Staphylococcus aureus. Front Cell Infect Microbial. 2017;7:286.
Crossref - Pietrocola G, Nobile G, Rindi S, Speziale P. Staphylococcus aureus manipulates innate immunity through own and host-expressed proteases. Front Cell Infect Microbiol. 2017;7:166.
Crossref - van Kessel KPM, Bestebroer J, van Strijp JAG. Neutrophil-mediated phagocytosis of Staphylococcus aureus. Front Immunol. 2014;5:467.
Crossref - Schmidt CQ, Lambris JD, Ricklin D. Protection of host cells by complement regulators. Immunol Rev. 2016;274(1):152-171.
Crossref - Burman JD, Leung E, Atkins KL, et al. Interaction of human complement with Sbi, a staphylococcal immunoglobulin-binding protein indications of a novel mechanism of complement evasion by Staphylococcus aureus. J Biol Chem. 2008;283(25):17579-17593.
Crossref - Koch TK, Reuter M, Barthel D, et al. Staphylococcus aureus proteins Sbi and Efb recruit human plasmin to degrade complement C3 and C3b. PLoS One. 2012;7(10):e47638.
Crossref - Uribe-Querol E, Rosales C. Control of phagocytosis by microbial pathogens. Front Immunol. 2017;8:1368.
Crossref - Gordon S. Phagocytosis: an immunobiologic process. Immun. 2016;44(3):463-475.
Crossref - Cockx M, Gouwy M, van Damme J, Struyf S. Chemoattractants and cytokines in primary ciliary dyskinesia and cystic fibrosis: key players in chronic respiratory diseases. Cell Molec Immunol. 2018;15(4):312-323.
Crossref - Cunha AF, Andrade HM, Souza FN, et al. Comparison of antibody repertories against Staphylococcus aureus in healthy and infected dairy cows with a distinct mastitis history and vaccinated with a polyvalent mastitis vaccine. J Dairy Sci. 2020;103(5):4588-4605.
Crossref - Cutone A, Lepanto MS, Rosa L, et al. Aerosolized bovine lactoferrin counteracts infection, inflammation and iron dysbalance in a cystic fibrosis mouse model of Pseudomonas aeruginosa chronic lung infection. Int J Molec Sci. 2019;20(9):2128.
Crossref - McGuinness WA, Kobayashi SD, DeLeo FR. Evasion of neutrophil killing by Staphylococcus aureus. Pathog. 2016;5(1):32.
Crossref - Flannagan RS, Heit B, Heinrichs DE. Antimicrobial mechanisms of macrophages and the immune evasion strategies of Staphylococcus aureus. Pathog. 2015;4(4):826-868.
Crossref - Shannon O, Nordenfelt P. Measuring antibody orientation at the bacterial surface. Methods Molec Biol. 2017;1535:331-337.
Crossref - Koenderman L. Inside-out control of Fc-receptors. Front immunol. 2019;10:971.
Crossref - Foster TJ, Geoghegan JA, Ganesh VK, Hook M. Adhesion, invasion and evasion: the many functions of the surface proteins of Staphylococcus aureus. Nat Rev Microbiol. 2014;12(1):49-62.
Crossref - Li W, Ulm H, Rausch M, et al. Analysis of the Staphylococcus aureus capsule biosynthesis pathway in vitro: characterization of the UDP-GlcNAc C6 dehydratases CapD and CapE and identification of enzyme inhibitors. Int J Med Microbiol. 2014;304(8):958-969.
Crossref - Oliveira D, Borges A, Simoes M. Staphylococcus aureus toxins and their molecular activity in infectious diseases. Toxins. 2018;10(6):252.
Crossref - Otto M. Staphylococcus aureus toxins. Curr Opin Microbiol. 2014;17:32-37.
Crossref - Astley R, Miller FC, Mursalin MH, Coburn PS, Callegan MC. An eye on Staphylococcus aureus toxins: roles in ocular damage and inflammation. Toxins. 2019;11(6):356.
Crossref - Pauli NT, Kim HK, Falugi F, et al. Staphylococcus aureus infection induces protein A-mediated immune evasion in humans. J Exper Med. 2014;211(12):2331-2339.
Crossref - Karauzum H, Datta SK. Adaptive immunity against Staphylococcus aureus. Curr Top Microbiol Immunol. 2017;409:419-439.
Crossref - Dunyach-Remy C, Ngba Essebe C, Sotto A, Lavigne JP. Staphylococcus aureus toxins and diabetic foot ulcers: role in pathogenesis and interest in diagnosis. Toxins. 2016;8:209.
Crossref - Liesenborghs L, Verhamme P, Vanassche T. Staphylococcus aureus, master manipulator of the human hemostatic system. J Thromb Haemost. 2018;16(3):441-454.
Crossref - Kwiecinski JM, Horswill AR. Staphylococcus aureus bloodstream infections: pathogenesis and regulatory mechanisms. Curr Opin Microbiol. 2020;53:51-60.
Crossref - Crosby HA, Kwiecinski J, Horswill AR. Staphylococcus aureus aggregation and coagulation mechanisms and their function in host-pathogen interactions. Adv Appl Microbiol. 2016;96:1-41.
Crossref - Paharik AE, Horswill AR. The staphylococcal biofilm: adhesins, regulation, and host response. Microbiol Spectr. 2016;4:529-566.
Crossref - Thammavongsa V, Kim HK, Missiakas D, Schneewind O. Staphylococcal manipulation of host immune responses. Nat Rev Microbiol. 2015;13(9):529-543.
Crossref - Broker BM, Mrochen D, Peton V. The T cell response to Staphylococcus aureus. Pathog. 2016;5(1):31.
Crossref - Hess C, Kemper C. Complement-mediated regulation of metabolism and basic cellular processes. Immun. 2016;45(2):240-254.
Crossref - Thakur A, Mikkelsen H, Jungersen G. Intracellular pathogens: host immunity and microbial persistence strategies. J Immunolo Res. 2019;2019:1356540.
Crossref
© The Author(s) 2020. Open Access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License which permits unrestricted use, sharing, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.