


ISSN: 0973-7510
E-ISSN: 2581-690X
Vibrio parahaemolyticus is a Gram-negative bacterium that is ubiquitous in marine and estuarine environments. The pathogenic strains can cause foodborne gastroenteritis especially when raw or undercooked seafood contaminated with pathogenic V. parahaemolyticus are consumed. This study aims to obtain a novel species-specific genetic marker using comparative genomics and to investigate their prevalence in both clinical and seafood isolates of V. parahaemolyticus. VPA1327 (vopT) was identified as a unique gene of this organism, but it was reported present in only 33.3% of V. parahaemolyticus (n=72 isolates). Interestingly, vopT was found exclusively in clinical isolates. The combination PCR analysis was used to determine the presence of three genes (tdh, trh, and vopT) in 36 clinical isolates. Four genotypes were classified namely, tdh+trh–vopT + (24 isolates, 66.7%), tdh+trh+vopT – (2 isolates, 5.6%), tdh–trh+vopT – (2 isolates, 5.6%) and tdh–trh–vopT– (8 isolates, 22.2%). Furthermore, all vopT+ isolates were tdh+ isolates, and none of them coexist with trh+ isolates. This study has provided strong evidence to support vopT as an additional virulence marker for the detection of V. parahaemolyticus with the pathogenic potential to cause human disease.
Vibrio parahaemolyticus, comparative genomic, VPA1327, VopT, T3SS, TDH
Vibrio parahaemolyticus is a Gram-negative halophilic marine and estuarine bacterium, which is an important pathogen causing seafood-borne gastroenteritis and occasional outbreaks1. This microorganism is generally detected in various seafood including oyster, clam, shrimp, and fish. Thus, consumption of raw or undercooked seafood contaminated with a virulent strain may put the individual at risk of getting the infection and causing disease. Detection and identification of V. parahaemolyticus strains in clinical and food samples are essential for diagnosing the cause of disease, as well as for surveillance and risk assessment of the contaminated food. High specificity and sensitivity detection assay are needed. For this purpose, polymerase chain reaction (PCR), a molecular DNA-based method has been developed and widely used since it is a rapid, convenient and applicable assay. The specificity of the test usually depends on the uniqueness of the target DNA (V. parahaemolyticus). Till date, toxR2 and tl3 are widely used for specific detection of V. parahaemolyticus4–7. However, some strains revealed false positive or false negative results to these genes in PCR assay5 due to high diversities and its close relation with Vibrio species. Therefore, a novel genetic marker with higher specificity is required for reliable diagnosis. Besides accurately identifying the organisms, their pathogenic potential also need to be evaluated. Most of V. parahaemolyticus strains isolated from diarrheal patients carry genes encoding thermostable direct hemolysin (TDH) and/or TDH-related hemolysin (TRH). TDH, but not TRH is responsible for the Kanagawa phenomenon (KP) showing beta hemolysis on a particular blood agar plate (Wagatsuma agar)8,9. However, it was demonstrated that pathogenicity of V. parahaemolyticus is not exclusively the responsibility of TDH10 and TRH11.
KP-positive V. parahaemolyticus strain, RIMD2210633, has two chromosomes, chromosome 1 (large) and chromosome 2 (small). The large chromosome contains most of the essential genes required for growth and viability, whereas, the small chromosome play a role in adaptation to environmental changes12. Besides being smaller in size, the chromosome 2 is more diverse in genetic organization and content. Genome sequencing data also revealed the presence of two sets of type III secretion system (T3SS) genes located in chromosome 1 (T3SS1) and 2 (T3SS2). The T3SS2 which is more divergent from the T3SS in gram-negative bacteria is present only in pathogenic KP-positive strains12. T3SS1 is responsible for cytotoxicity, whereas, T3SS2 is responsible for enterotoxicity and intestinal fluid accumulation13. T3SS2 was further characterized and two distinct lineages, T3SS2a and T3SS2b were further classified13–15. T3SS2a is associated with tdh (tdh+ trh–) isolates, while T3SS2b is associated with trh (tdh– trh+) isolates.
As earlier stated, the success of genome sequencing has contributed widely to related genomic studies especially the comparative genomic studies. By using in silico comparison, specific targets or genetic markers for the detection and identification of pathogenic bacteria can be identified7,16–18. This study primarily aimed to explore a novel genetic marker for exclusively detected V. parahaemolyticus by comparison analysis between chromosome 2 of V. parahaemolyticus and V. alginolyticus.
Bacterial strains
A total of 125 bacterial strains used in this study were collected and kept in our laboratory. There were 106 and 19 stains of Vibrionaceae and non-Vibrionaceae, respectively. Among the Vibrionaceae, 72 strains were V. parahaemolyticus including ATCC17802 (1), clinical (36) and seafood (35) strains and 34 strains were non-V. parahaemolyticus. All bacterial strains and their sources were listed in Table 1.
Table (1):
Bacterial strains used in this study.
Bacterial species |
No. of strains |
Source (number of isolates) |
|---|---|---|
V. parahaemolyticus |
72 |
ATCC17802 (1), clin (36), sf (35) |
V. alginolyticus |
10 |
ATCC17749 (1), clin (5), sf (4) |
V. cholerae |
5 |
Ref. (3 i.e., N16961, 569B, O17SR. ), clin (1), sf (1) |
V. fluvialis, |
5 |
Ref.V26/36 (1), ), |
V. furnisii |
3 |
clin (3) clin (3), sf (1) |
V. vulnificus |
2 |
ATCC27526 (1), sf (1) |
V. mimicus |
3 |
clin (1), sf (2) |
V. harveyi |
2 |
ATCC14126 (1), sf (1) |
V. campbellii |
1 |
ATCC25920 (1) |
Grimontia hollisae |
1 |
ATCC33564 (1) |
Photobacterium damselae, |
2 |
sf (2) |
Providencia rettigeri |
1 |
unk |
Escherichia coli |
1 |
ATCC25933 |
Pseudomonas aeruginosa |
4 |
ATCC27857, Ref. (3 i.e., DMS00554, L165NC, P137) |
Shigella flexneri |
3 |
clin (3) |
Salmonella Typhimurium |
1 |
ATCC13311 (1) |
S. Enteritidis |
1 |
clin |
S. Paratyphi A |
1 |
clin |
S. Oslo |
1 |
unk |
S. Orion |
1 |
unk |
Klebsiella pneumoniae |
1 |
unk |
Citrobacter fruendii |
1 |
unk |
Enterobacter cloacae |
1 |
unk |
Enterococcus faecalis |
1 |
unk |
Edwardsiella tarda |
1 |
unk |
ATCC, American Type Culture Collection; clin, clinical isolate; sf, seafood isolate; unk, unknown origin
Comparison between chromosome II genome sequences of V. parahaemolyticus and V. alginolyticus
Closely related chromosome II DNA sequences of V. parahaemolyticus RIMD2210633 (Accession No. NC_004605.1) and V. alginolyticus ATCC 17749 (Accession no. NC_022359.1) were retrieved from the GeneBank database. Firstly, the two genome sequences were aligned using Microbial Nucleotide BLAST and BLASTn from Center for Biotechnology Information (http://blast.ncbi.nlm.nih.gov/) optimized to produce highly similar sequences. The uniquely identified regions on V. parahaemolyticus chromosome II were then selected and analyzed for their gene identifications (ID) and corresponding coding sequences (CDSs) were obtained from the database (https://www.ncbi.nlm.nih.gov/nuccore). To confirm the absence of the archive V. parahaemolyticus-specific sequence, these CDSs were individually aligned against non-redundant protein sequence (nr) using BLASTx with the exclusion of V. parahaemolyticus sequences. The CDSs revealed no similarity/identity with other Vibrio spp. ORFs or low identity to the proteins of other genus was considered as unique genes of V. parahaemolyticus.
The primers of the selected unique CDS were designed using DNAMAN (Lynnon BioSoft, Canada). All primers used in this study were commercially synthesized (Bioneer, South Korea).
Preparation of DNA template
A colony of the bacterial strains cultured overnight was picked, suspended in sterile distilled water (DW) and the OD600nm was adjusted to one. After the adjusted bacterial cell was boiled and centrifuged at 8,100 xg for 5 min, the supernatant was transferred to a new tube and kept at 4°C for use as a template for PCR amplification.
PCR amplification of vpa1327
The VPA1327 primers were shown in Table 2. The reaction mixture contained 1.0 U of Taq polymerase (Fermentas, USA), 1x of reaction buffer containing KCl, 2.0 mM MgCl2, 0.4 µM of each primer vpa1327F and vpa1327R, 0.2 mM of each deoxynucleoside triphosphate (dNTP), 1 µl of bacterial DNA template and the reaction volume which was made up to 20 µl using sterile DW. The PCR cycling conditions were one cycle of 5 min at 95°C followed by 30 cycles of 30 s at 95°C, 30 s at 58°C and 30 s at 72°C followed by a final extension time of 5 min at 72°C. The PCR products were electrophoresed in 1.5% agarose gel, stained with ethidium bromide, and then visualized by a gel documentation system (Syngene, UK).
Table (2):
Sequences of primers used in this study.
Gene |
Primer sequences |
Size (bp) |
Reference |
|---|---|---|---|
toxR |
F: 5′-GTCTTCTGACGCAATCGTTG-3′ R: 5′-ATACGAGTGGTTGCTGTCATG-3′ |
368 |
Kim et al.2 |
tdh |
F: 5′-GTAAAGGTCTCTGACTTTTGGAC-3′ R: 5′-TGGAATAGAACCTTCATCTTCACC-3′ |
269 |
Bej et al.3 |
trh |
F: 5′-CGCTCTCATATGCTTCGAC-3′ R: 5′-TTCGCGATTGATCTACCA-3′ |
433 |
this study |
vpa1327 |
F: 5′-ACATACGGAAAATATAGGTAGTG-3′ R: 5′-AAATCTAGCGCATCAAGT -3′ |
687 |
this study |
16SrRNA |
F: 5′-CGGTGAAATGCGTAGAGAT -3′ R: 5′-TTACTAGCGATTCCGAGTTC -3′ |
663 |
Tarr et al.33 |
F, Forward primer; R, Reverse primer.
Multiplex PCR
The multiplex PCR was designed to determine toxR, tdh and trh of V. parahaemolyticus strains within one reaction tube only. The reaction mixture volume of 25 µl contained 1.0 µl of DNA template, 1.0 U Taq DNA polymerase, 1x Taq buffer containing KCl, 2.0 mM MgCl2, 0.2 mM for each dNTPs, and 0.4, 0.8, 0.4 and 0. 2 mM of tdh, trh, toxR and 16SrRNA primer pair respectively (Table 2). The thermal cycling was set similarly for vpa1327 amplification. The PCR products were examined by agarose gel electrophoresis as described in the previous section.
This study primarily aimed to identify a unique gene encoding for a protein that is unique to V. parahaemolyticus by genomic comparison. To identify the unique gene, the ‘in silico’ genomic comparison between chromosome II DNA sequences of V. parahaemolyticus (1,877,212 bp) and closely related species, as well as that of V. alginolyticus (1,812,170 bp) was done. The reason behind the usage of chromosome II for comparison is that it is more diverse in structure and gene content than chromosome I. Generally, 56.8% unique genes of V. parahaemolyticus were carried from chromosome II, whereas, only 29.5% were carried from chromosome I12. Moreover, genes encoding type III secretion system (T3SS2) located on this chromosome was previously considered as being related to the pathogenicity of V. parahaemolyticus to humans15. Chromosomes II from both species were compared based on nucleotide sequence homology using BLASTn, and the BLASTn analysis showed 94% homology over 64% coverage of chromosome II nucleotide sequence. Approximately 200 discrete regions located on the small chromosome of V. parahaemolyticus were predicted as the region of differences. Among these, 106 regions were more extended than 1,000 bp.
The three large regions spanning over 20,000 bp were located, and their predicted CDSs were retrieved from V. parahaemolyticus genome sequence (Accession No. NC_004605.1) in the NCBI database as shown and summarized in Fig. 1. A total of 121 CDSs were further analyzed by BLASTx, and the results indicated that only two designated genes, VPA1327 and VPA1331, revealing low identity to the proteins of other genus were selected. VPA1327 encoding an exoenzyme T revealed 64% (over 233 amino acids) and 46 % (over 173 amino acids) with exoenzyme T of Providencia alcalifaciens and Pseudomonas aeruginosa, respectively. Interestingly, this orthologous exoenzyme T (VPA1327) was not found in Vibrio spp. other than V. parahaemolyticus. In contrast, VPA1331 exhibited one matched sequence with 41% identity to the putative OspC2 of Vibrio spp. strain FF_286. These results indicated that VPA1327 is unique to V. parahaemolyticus CDS.
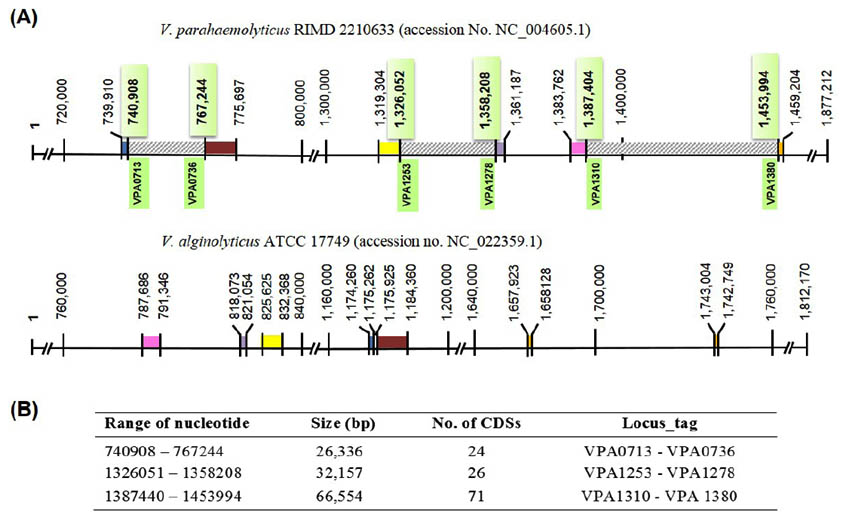
Fig. 1. Comparative chromosomal 2 genome V. parahaemolyticus RIMD 2210633 (accession No. NC_004605.1) and V. alginolyticus ATCC 17749 (accession no. NC_022359.1). (A), Diagram of V. parahaemolyticus chromosome 2 (upper panel) showed three selected regions of differences (RD, striped boxes) which were not found on the chromosome 2 of V. alginolyticus (lower panel). The flanking regions of each RD are homologous regions shown as identically colored blocks in each genome. (B), List of three selected RD and their CDSs carried on V. parahaemolyticus chromosome 2.
Since VPA1327 gene encoding for an exoenzyme T, also known as Vibrio outer protein (Vop) T was found to be unique to V. parahaemolyticus, we hypothesized the use of vopT as a genetic marker for specific detection. Although, there are several genes used for diagnosis of V. parahaemolyticus such as toxR2, tl3 and atpA19, they were not uniquely designed for V. parahaemolyticus. These gene sequences, 86, 97, and 85% respectively, showed a high similarity with those of V. alginolyticus7. As previously described in our study that apart from V. parahaemolyticus, vopT (VPA1327) was not found in vibrios, therefore, exclusive specificity should be obtained from V. parahaemolyticus detection based on a vopT gene. To detect the vopT, the specific primers were designed. The PCR assay was developed, and the optimal annealing temperature was determined to be 58°C. The PCR reaction yielded an amplified fragment of 687 bp. The specificity of the designed primer pair was evaluated by the optimized single PCR using various Vibrio species and other representative pathogenic bacteria, as listed in Table 1. The PCR results showed that vopT was specific to V. parahaemolyticus, deprived of false positive submitting to 100% specificity as expected. Nevertheless, only 24 of 72 (33.3%) tested V. parahaemolyticus strains revealed positive results reflecting vastly low sensitivity to accept detection application. Interestingly, these all positive strains were from clinical samples. This observation prompted us to determine whether vopT could be used to distinguish between pathogenic strains from nonpathogenic ones. Therefore, all V. parahaemolyticus strains used in our study were further used to determine the presence of toxR, tdh, and trh using optimized multiplex PCR.
Table (3):
Distribution of tdh, trh, and vopT in clinical isolates of V. parahaemolyticus by PCR.
| No. | V. parahaemolyticus isolate |
PCR result |
||||
|---|---|---|---|---|---|---|
| 16SrRNA | toxR | tdh | trh | vopT | ||
| 1 | 52605281 | + | + | + | – | + |
| 2 | 53607188 | + | + | + | – | + |
| 3 | 52605165 | + | + | + | – | + |
| 4 | 52605981 | + | + | + | – | + |
| 5 | 52604921 | + | + | + | – | + |
| 6 | 52605516 | + | + | + | – | + |
| 7 | 52604579 | + | + | + | – | + |
| 8 | 52604517 | + | + | – | – | – |
| 9 | 52605668 | + | + | – | – | – |
| 10 | 52605556 | + | + | + | – | + |
| 11 | 52605517 | + | + | – | – | – |
| 12 | 52605675 | + | + | – | – | – |
| 13 | 52605774 | + | + | + | – | + |
| 14 | 52605822 | + | + | + | – | + |
| 15 | 53-01 | + | + | + | – | + |
| 16 | 53-02 | + | + | + | – | + |
| 17 | 53-04 | + | + | + | – | + |
| 18 | 53-05 | + | + | + | – | + |
| 19 | 53-06 | + | + | + | – | + |
| 20 | 53-07 | + | + | – | – | – |
| 21 | 53603951 | + | + | + | – | + |
| 22 | 53603910 | + | + | + | – | + |
| 23 | 53604316 | + | + | + | – | + |
| 24 | 53604513 | + | + | – | – | – |
| 25 | 53604518 | + | + | + | – | + |
| 26 | 53605975 | + | + | + | + | – |
| 27 | 53606206 | + | + | – | – | – |
| 28 | 53606637 | + | + | + | – | + |
| 29 | 53607058 | + | + | + | – | + |
| 30 | 53608005 | + | + | – | + | – |
| 31 | 53609903 | + | + | + | – | + |
| 32 | 53610963 | + | + | + | – | + |
| 33 | 53611025 | + | + | – | + | – |
| 34 | 53611080 | + | + | – | – | – |
| 35 | 53611224 | + | + | + | – | + |
| 36 | 53607926 | + | + | + | + | – |
| 37 | ATCC17802 | + | + | – | + | – |
All 106 Vibrionaceae were positive for 16SrRNA of which 72 isolates were toxR+, species-specific marker confirmed the culture-based identification of V. parahaemolyticus used in this study. A total of 35 seafood isolates were negative for both tdh and trh indicating nonpathogenic V. parahaemolyticus, of which vopT was also absent. In contrast, 26 (72.2%), 4 (11.1%) and 24 (66.7%) of 36 clinical isolates besides ATCC17802 were positive for tdh, trh, and vopT, respectively (Table 3). These results are consistent with several previous studies as summarized in Table 4. These studies showed quite high frequency of tdh which is more than 50% of the clinical isolates. However, contradicting results reported by Jone et al.20 showed a higher prevalence of trh (61.7%) but a quite low proportion of vopT (11.7%) in clinical isolates. The difference may be due to differences in geographic regions where the bacterium was isolated. The Jone’s study was carried out in USA20, while others were carried out in Asian Countries such as China21–23 and Thailand24, including this study. Additionally, the PCR results of the three genes of clinical isolates in this study could be classified into four groups (Table 4) i.e., tdh+trh–vopT + (24 isolates), tdh+trh+vopT – (2 isolates), tdh–trh+vopT – (2 isolates) and tdh–trh–vopT– (8 isolates). These results showed that all vopT+ isolates were tdh+, accounted for 92.3% (24/26) and none of them coexist with trh. The vopT express exclusively with tdh, as demonstrated in several studies13,14,20,22,25 including this study. However, few isolates lack tdh, with or without trh, but express vopT could be found as reported recently21,24.
Table (4):
The occurrence of tdh, trh, and vopT in clinical V. parahaemolyticus isolates compared to data retrieved from several studies.
| Gene | This study % (no.) |
Jones et al.20 % (no.) |
Tsai et al.22 % (no.) |
Mala et al.24 % (no.) |
Chen et al.23 % (no.) |
Li et al.21 % (no.) |
|---|---|---|---|---|---|---|
| no. isolates | 36 | 77 | 49 | 74 | 501 | 42 |
| tdh | 72.2% (26) | 57.1% (44) | 85.7% (42) | 93.2% (69) | 93.0% (466) | 88.1% (37) |
| trh | 11.1% (4) | 61.0% (47) | 2.0% (1) | 10.8% (8) | 1% (5) | 16.7% (7) |
| vopT | 66.7% (24) | 11.7% (9) | 85.7% (42) | 98.6% (73) | 93.0% (466) | 90.5% (38) |
| vopB2 | nd | 11.7% (9) | 85.7% (42) | 89.2% (66) | 93.0% (466) | 95.2% (40) |
| Gene combination | ||||||
| tdh+trh–vopT+ | 66.7% (24) | 11.7% (9) | na | 70.8% (17/24*) | 92.4% (463) | 80.9% (34) |
| tdh+trh+vopT+ | 0% (0) | 0% (0) | na | 20.8% (5/24*) | 0.6% (3) | 2.4% (1) |
| tdh–trh+vopT+ | 0% (0) | 0% (0) | na | 4.2% (1/24*) | 0% (0) | 4.8% (2) |
| tdh–trh–vopT+ | 0% (0) | 0% (0) | na | 4.2% (1/24*) | 0% (0) | 2.4% (1) |
| tdh–trh–vopT– | 22.2% (8) | 27.3% (21) | na | 0% (0/24*) | 6.6% (33) | 0% (0) |
nd, not determined; na, not available; *, data provided for only 24 samples
TDH and TRH encoded by tdh and trh genes, respectively, are considered as major virulence factors in V. parahaemolyticus26,27. It was found that the majority of clinical cases were associated with V. parahaemolyticus strains carrying either tdh or trh8,20, and they are therefore used as indicators for the pathogenic strains. Our study also revealed that most of the clinical isolates of V. parahaemolyticus (28/36, 77.8%) carried either tdh or trh. Among 28 isolates, almost all isolates (26, 72.2%) were tdh+, but only 4 (11.1%) trh+ were in good agreement with several previous studies reported. For example, the distribution of tdh and trh in clinical isolates were respectively 63.6% vs. 35.5%8 and 84% vs. 12%25. These finding indicated that trh gene is less pronounced for pathogenic V. parahaemolyticus isolated from clinical samples. In contrast, trh positive strains were more frequently isolated from the environment or seafood than tdh positive ones, e.g., only 7% (5/71) of environmental strains carry trh gene, while tdh gene could not be detected8. Vongxay et al.25 also reported that 1.6% and 3.7% of 191 seafood samples carried tdh and trh, respectively. Within the human host, the hostile environment in the intestine, i.e., high temperature as well as the presence of bile is a potent stimulator to upregulate the expression of genes encoding TDH and T3SS2 including vopT28. Therefore, tdh gene was detected more frequently in clinical than in seafood or environmental strains. In our study, none of tdh or trh was found in all 35 V. parahaemolyticus strains from seafood samples. Since the prevalence of pathogenic strains in the environment or seafood are very low, there is minimal opportunity for tdh/trh genes to recover unless the higher number of isolates are examined. Although, either tdh or trh is indicated as genetic markers for the pathogenic strains, 22.2% of clinical strains lacking both tdh and trh (tdh–trh–) were obtained in this study similar to those obtained in several previous studies8,20. Additionally, it was found that deletion of tdh from trh-negative strain merely partially inhibited cytotoxicity10. This evidence indicated that the two virulence factors, TDH and TRH, do not adequately account for V. parahaemolyticus pathogenicity and suggested the existence of other virulence factors besides TDH and TRH20,29. Owing to recent report that vopT could be detected in clinical isolates lacking tdh, trh or both genes21,24, VopT may result in the pathogenicity of these strains.
Kodama and colleagues30 had identified and characterized VopT and reported that it is one of the T3SS2 effector proteins which play a role in enterotoxicity. The amino acid sequence of VopT revealed 45% and 44% identity with the C-terminal ADP-ribosyltransferase (ADPRT) domain of two effector proteins secreted by T3SS of P. aeruginosa, exoenzyme T, and exoenzyme S, respectively. The ADPRT domain of exoenzyme T was shown to involve in the pathogenesis of P. aeruginosa infection31. Also, VopT was previously30 shown to be secreted and injected through T3SS2 into the cytoplasm of the host cells in which Ras protein was ADP-ribosylation. Like exoenzyme T of P. aeruginosa, the ADPRT domain of VopT is required for cytotoxicity. The cytotoxic activity decreased when vopT was deleted. Hence, it was implied that VopT is one of the virulence factors produced by the organism and it plays an important role in inducing cytotoxicity. Moreover, our results showed that vopT was detected mainly from the majority of clinical isolates (66.7%) and coexisted consistently with tdh (tdh+vopT+, 92.3%). Therefore, the expression of vopT is tdh or tdh-dependent. However, it may function independently for the pathogenicity of a few isolates harboring vopT without either tdh or trh. From the study of V. parahaemolyticus pathogenicity, it was observed that the enteropathogenicity of trh-positive isolates seemed to fall between tdh-positive isolates and those without tdh and trh25. Likewise, the presence or absence of vopT may reflect the degree of virulence or pathogenicity of V. parahaemolyticus. VopT may function parallel to or in agreement with TDH which need further investigation.
The distribution of vopT among the clinical isolates revealed in this study was similar to that of vopB2, another effector gene of T3SS2a, reported by Noriea et al.14. Since vopB2 was found only in the clinical isolates and co-existed with tdh gene but was not found in the environmental strains, Noriea et al. suggested vopB2 to be a representative gene for identifying increased virulence among strains. However, the recent studies revealed that the three genes, i.e., tdh, vopT and vopB2 were not coexisting stringently and few vopT/vopB2 gene could be found together with trh-positive or tdh-negative isolates21,23,24. By comparing the detection rate of vopT and vopB2 genes among several studies, the positive rate of vopT is usually equal to vopB2 since they coexist, but one study found the percentage of vopT over vopB2 and vice versa in another study (Table 4). Moreover, VopT was found unique to V. parahaemolyticus in opposition to TDH which is a major virulence factor and is not restricted to only V. parahaemolyticus, but it has also been documented in other Vibrionaceae such as G. hollisae, V. mimicus, and V. cholerae32. Taken together, we considered that vopT, likewise tdh, is also a major virulence factor and the use of vopT was suggested, in addition to tdh and vopB2, as a genetic marker for more reliable identification of pathogenic V. parahaemolyticus.
The objective of the present study was primarily to search for a new genetic marker for the identification of V. parahaemolyticus by genomic analysis approach. Finally, our study has identified vopT (VPA1327), which is one of the T3SS2 effector genes specific to V. parahaemolyticus. The distribution of vopT is the same as tdh, and being mainly obtained from clinical isolates indicated that they were associated with pathogenicity. Moreover, vopT always coexist with tdh, but it is not dependent on tdh. Therefore, in this study, we proposed the screening of vopT parallel to tdh for more reliable identification of pathogenic V. parahaemolyticus strains.
ACKNOWLEDGMENTS
This research was supported by National Research Council of Thailand and Health Systems Research Institute.
- Nair GB, Ramamurthy T, Bhattacharya SK, Dutta B, Takeda Y, Sack DA. Global dissemination of Vibrio parahaemolyticus serotype O3:K6 and its serovariants. Clin Microbiol Rev. 2007; 20(1):39-48. doi:10.1128/CMR.00025-06.
- Kim YB, Okuda J, Matsumoto C, Takahashi N, Hashimoto S, Nishibuchi M. Identification of Vibrio parahaemolyticus strains at the species level by PCR targeted to the toxR gene. J Clin Microbiol. 1999; 37(4):1173-1177.
- Bej AK, Patterson DP, Brasher CW, Vickery MCL, Jones DD, Kaysner CA. Detection of total and hemolysin-producing Vibrio parahaemolyticus in shellfish using multiplex PCR amplification of tl, tdh and trh. J Microbiol Methods. 1999; 36(3):215-225. doi:10.1016/S0167-7012(99)00037-8.
- Caburlotto G, Gennari M, Ghidini V, Tafi M, Lleo MM. Presence of T3SS2 and other virulence-related genes in tdh-negative Vibrio parahaemolyticus environmental strains isolated from marine samples in the area of the Venetian Lagoon, Italy. FEMS Microbiol Ecol. 2009; 70(3):506-514. doi:10.1111/j.1574-6941.2009.00764.x.
- Croci L, Suffredini E, Cozzi L, et al. Comparison of different biochemical and molecular methods for the identification of Vibrio parahaemolyticus. J Appl Microbiol. 2007; 102(1):229-237. doi:10.1111/j.1365-2672.2006.03046.x.
- Jiang Y, He L, Wu P, et al. Simultaneous identification of ten bacterial pathogens using the multiplex ligation reaction based on the probe melting curve analysis. Sci Rep. 2017; 7(1):1-9. doi:10.1038/s41598-017-06348-z.
- Li R, Chiou J, Chan EWC, Chen S. A novel PCR-based approach for accurate identification of Vibrio parahaemolyticus. Front Microbiol. 2016; 7(JAN):1-5. doi:10.3389/fmicb.2016.00044.
- Shirai H, Ito H, Hirayama T, et al. Molecular epidemiologic evidence for association of thermostable direct hemolysin (TDH) and TDH-related hemolysin of Vibrio parahaemolyticus with gastroenteritis. Infect Immun. 1990; 58(11):3568-3573.
- Honda T, Iida T. The pathogenicity of Vibrio parahaemolyticus and the role of the thermostable direct haemolysin and related haemolysins. Rev Med Microbiol. 1993; 4(2):106-113. doi:10.1097/00013542-199304000-00006.
- Park KS, Ono T, Rokuda M, Jang MH, Iida T, Honda T. Cytotoxicity and enterotoxicity of the thermostable direct hemolysin-deletion mutants of Vibrio parahaemolyticus. Microbiol Immunol. 2004; 48(4):313-318. doi:10.1111/j.1348-0421.2004.tb03512.x.
- Ming X, Yamamoto K, Honda T. Construction and characterization of an isogenic mutant of Vibrio parahaemolyticus having a deletion in the thermostable direct hemolysin- related hemolysin gene (trh). J Bacteriol. 1994; 176(15):4757-4760.
- Makino K, Oshima K, Kurokawa K, et al. Genome sequence of Vibrio parahaemolyticus: a pathogenic mechanism distinct from that of V. cholerae. Lancet. 2003; 361(9359):743-749. doi:10.1016/S0140-6736(03)12659-1.
- Park K-S, Ono T, Rokuda M, et al. Functional characterization of two type III secretion systems of Vibrio parahaemolyticus. Infect Immun. 2004; 72(11):6659-6665. doi:10.1128/IAI.72.11.6659-6665.2004.
- Noriea NF, Johnson CN, Griffitt KJ, Grimes DJ. Distribution of type III secretion systems in Vibrio parahaemolyticus from the northern Gulf of Mexico. J Appl Microbiol. 2010; 109(3):953-962. doi:10.1111/j.1365-2672.2010.04722.x.
- Okada N, Iida T, Park KS, et al. Identification and characterization of a novel type III secretion system in trh-positive Vibrio parahaemolyticus strain TH3996 reveal genetic lineage and diversity of pathogenic machinery beyond the species level. Infect Immun. 2009; 77(2):904-913. doi:10.1128/IAI.01184-08.
- Amaral GRS, Dias GM, Wellington-Oguri M, et al. Genotype to phenotype: Identification of diagnostic Vibrio phenotypes using whole genome sequences. Int J Syst Evol Microbiol. 2014; 64(PART 2):357-365. doi:10.1099/ijs.0.057927-0.
- Kim H-J, Ryu J-O, Lee S-Y, Kim E-S, Kim H-Y. Multiplex PCR for detection of the Vibrio genus and five pathogenic Vibrio species with primer sets designed using comparative genomics. BMC Microbiol. 2015; 15(1):239. doi:10.1186/s12866-015-0577-3.
- Yu S, Chen W, Wang D, He X, Zhu X, Shi X. Species-specific PCR detection of the food-borne pathogen Vibrio parahaemolyticus using the irgB gene identified by comparative genomic analysis. FEMS Microbiol Lett. 2010; 307(1):65-71. doi:10.1111/j.1574-6968.2010.01952.x.
- Izumiya H, Matsumoto K, Yahiro S, et al. Multiplex PCR assay for identification of three major pathogenic Vibrio spp., Vibrio cholerae, Vibrio parahaemolyticus, and Vibrio vulnificus. Mol Cell Probes. 2011; 25(4):174-176. doi:10.1016/j.mcp.2011.04.004.
- Jones JL, Lüdeke CHM, Bowers JC, et al. Biochemical, serological, and virulence characterization of clinical and oyster Vibrio parahaemolyticus isolates. J Clin Microbiol. 2012; 50(7):2343-2352. doi:10.1128/JCM.00196-12.
- Li H, Tang R, Lou Y, Cui Z, Chen W, Hong Q. A Comprehensive epidemiological research for clinical Vibrio parahaemolyticus in Shanghai. 2017; 8(June):1-10. doi:10.3389/fmicb.2017.01043.
- Tsai S, Jong K-J, Tey YH, et al. Molecular characterization of clinical and environmental Vibrio parahaemolyticus isolates in Taiwan. Int J Food Microbiol. 2013; 165(1):18-26. doi:10.1016/j.ijfoodmicro.2013.04.017.
- Chen Y, Chen X, Yu F, et al. Serology, virulence, antimicrobial susceptibility and molecular characteristics of clinical Vibrio parahaemolyticus strains circulating in southeastern China from 2009 to 2013. Clin Microbiol Infect. 2016; 22(3):258.e9-258.e16. doi:10.1016/j.cmi.2015.11.003.
- Mala W, Alam M, Angkititrakul S, et al. Sero group, virulence , and molecular traits of Vibrio parahaemolyticus isolated from clinical and cockle sources in northeastern Thailand. Infect Genet Evol. 2016; 39:212-218. doi:10.1016/j.meegid.2016.01.006.
- Vongxay K, Wang S, Zhang X, et al. Pathogenetic characterization of Vibrio parahaemolyticus isolates from clinical and seafood sources. Int J Food Microbiol. 2008; 126(1-2):71-75. doi:10.1016/j.ijfoodmicro.2008.04.032.
- Honda T, Ni Y, Miwatani T. Purification and characterization of a hemolysin produced by a clinical isolate of Kanagawa phenomenon-negative Vibrio parahaemolyticus and related to the thermostable direct hemolysin. Infect Immun. 1988; 56(4):961-965.
- Nishibuchi M, Fasano A, Russell RG, Kaper JB. Enterotoxigenicity of Vibrio parahaemolyticus with and without genes encoding thermostable direct hemolysin. Infect Immun. 1992; 60(9):3539-3545.
- Gotoh K, Kodama T, Hiyoshi H, et al. Bile acid-induced virulence gene expression of Vibrio parahaemolyticus reveals a novel therapeutic potential for bile acid sequestrants. PLoS One. 2010; 5(10). doi:10.1371/journal.pone.0013365.
- Lynch T, Livingstone S, Buenaventura E, et al. Vibrio parahaemolyticus disruption of epithelial cell tight junctions occurs independently of toxin production. 2005; 73(3):1275-1283. doi:10.1128/IAI.73.3.1275.
- Kodama T, Rokuda M, Park KS, et al. Identification and characterization of VopT, a novel ADP-ribosyltransferase effector protein secreted via the Vibrio parahaemolyticus type III secretion system 2. Cell Microbiol. 2007; 9(11):2598-2609. doi:10.1111/j.1462-5822.2007.00980.x.
- Garrity-Ryan L, Shafikhani S, Balachandran P, et al. The ADP Ribosyltransferase domain of Pseudomonas aeruginosa exoT cContributes to its biological activities. Infect Immun. 2004; 72(1):546-558. doi:10.1128/IAI.72.1.546-558.2004.
- Bhowmik SK, Pazhani GP, Ramamurthy T. Phylogenetic and in silico functional analyses of thermostable-direct hemolysin and tdh-related encoding genes in Vibrio parahaemolyticus and other gram-negative bacteria. Biomed Res Int. 2014;2014. doi:10.1155/2014/576528.
- Tarr CL, Patel JS, Puhr ND, Sowers EG, Bopp CA, Strockbine NA. Identification of Vibrio isolates by a multiplex PCR assay and rpoB sequence determination. J Clin Microbiol. 2007; 45(1):134-140. doi:10.1128/JCM.01544-06.
© The Author(s) 2018. Open Access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License which permits unrestricted use, sharing, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.