


ISSN: 0973-7510
E-ISSN: 2581-690X
Arif Nur Muhammad Ansori1,2,3![]() , Viol Dhea Kharisma2,3,4, Sahal Sabilil Muttaqin5, Yulanda Antonius2,6 and Arli Aditya Parikesit7
, Viol Dhea Kharisma2,3,4, Sahal Sabilil Muttaqin5, Yulanda Antonius2,6 and Arli Aditya Parikesit7 ![]()
Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2), a novel coronavirus and the primary causative agent of coronavirus disease 2019 (COVID-19), first occurred in China and rapidly spread worldwide. The government of the Republic of Indonesia confirmed its first two cases of COVID-19 in March 2020. COVID-19 is a serious illness with no efficacious antiviral medication or approved vaccine currently available. Therefore, there is a need to investigate the genome of SARS-CoV-2. In this study, we characterized SARS-CoV-2 spike glycoprotein genes from Indonesia to investigate their genetic composition and variability. Overall, ten SARS-CoV-2 spike glycoprotein gene sequences retrieved from GenBank (National Center for Biotechnology Information, USA) and the GISAID EpiCoV database (Germany) were compared. We analyzed nucleotide variants and amino acid changes using Molecular Evolutionary Genetics Analysis (MEGA) X and analyzed gene similarity using the LALIGN web server. Interestingly, we revealed several specific mutation sites, however, there were no significant changes in the genetic composition of SARS-CoV-2 spike glycoprotein genes, when compared to the Wuhan-Hu-1 isolate from China. However, this is a preliminary study and we recommend that molecular epidemiology and surveillance programs against COVID-19 in Indonesia be improved.
Coronavirus, COVID-19, Genetic composition, Mutation, SARS-CoV-2
The Chinese government first reported a novel pneumonia-causing disease in Wuhan in December 20191. The causative agent was identified and named severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) by the International Committee on Taxonomy of Viruses (ICTV)2. This new virus has rapidly spread across China and to many other countries across the world, including Indonesia3-4. The World Health Organization has named the illness caused by SARS-CoV-2 as coronavirus disease 2019 (COVID-19)5.
According to an online interactive dashboard hosted by the Center for Systems Science and Engineering at Johns Hopkins University (Baltimore, USA), which tracks reported cases of COVID-19 in real-time6, more than 4 million people have been infected by SARS-CoV-2 worldwide, with more than 14 000 cases in Indonesia alone. Currently, there are three coronaviruses that cause illness in humans: severe acute respiratory syndrome coronavirus (SARS-CoV), Middle East respiratory syndrome coronavirus (MERS-CoV), and SARS-CoV-27.
Taxonomically, coronaviruses belong to the Coronaviridae family in the order Nidovirales, with examples in four distinct genera: Alphacoronavirus, Betacoronavirus, Deltacoronavirus, and Gammacoronavirus8. The structural proteins are encoded by four genes, specifically the envelope (E), nucleocapsid (N), membrane (M), and spike glycoprotein (S)7,9-10. Previous studies have shown that the spike glycoprotein plays a crucial role in binding to receptors on the host cell. Therefore, this protein is a key target for a number of antiviral therapies and a promising antigen for generating vaccines formulated against SARS-CoV, MERS-CoV, and SARS-CoV-2.
Molecular epidemiology research is a crucial tool in the surveillance of newly emerging and reemerging viruses11-12. Indonesia was the eighth country in Southeast Asia after Brunei, Cambodia, Malaysia, Myanmar, Singapore, Thailand, and Vietnam to report the whole-genome sequences of SARS-CoV-2 in the region. Both Callaway (2020) and Shang et al. (2020) have shown that vaccines are being developed against SARS-CoV-2 by various research groups worldwide13-14. Similarly, Al-Tawfiq (2020) has discussed other potential therapeutic options for COVID-1915 and both remdesivir and chloroquine are capable of effectively inhibiting SARS-CoV-2 in in vitro assays16. Despite these promising treatment options, COVID-19 remains a serious disease with no proven effective antiviral medication or approved vaccine available. Therefore, there is an urgent need to investigate the genome of SARS-CoV-2. In this study, we characterized SARS-CoV-2 spike glycoprotein genes from Indonesia in order to investigate their genetic composition and the similarity between different gene isolates.
SARS-CoV-2 Isolates
SARS-CoV-2 spike glycoprotein gene (3822 bp) sequences were obtained from GenBank (National Center for Biotechnology Information, USA) and the Global Initiative on Sharing All Influenza Data (GISAID) EpiCoV database (Germany) (Table 1).
Table (1):
SARS-CoV-2 isolates obtained from the GenBank and GISAID EpiCoV databases.
No |
Accession ID |
Virus Name |
Origin |
Submitting Institution |
Host |
Specimen Source |
Coverage |
|---|---|---|---|---|---|---|---|
1 |
MN908947.3 (Reference) |
Wuhan-Hu-1 |
China (Wuhan) |
Shanghai Public Health Clinical Center and School of Public Health, Fudan University, Shanghai |
Homo sapiens |
Unknown |
Reference genome |
2 |
EPI_ISL_435281 |
JKT-EIJK0141 |
Indonesia (Jakarta) |
Eijkman Institute for Molecular Biology, Ministry of Research and Technology/National Agency for Research and Innovation of the Republic of Indonesia |
Homo sapiens |
Nasopharyngeal and Oro-pharyngeal swab |
22× |
3 |
EPI_ISL_435282 |
JKT-EIJK0317 |
Indonesia (Jakarta) |
Eijkman Institute for Molecular Biology, Ministry of Research and Technology/National Agency for Research and Innovation of the Republic of Indonesia |
Homo sapiens |
Nasopharyngeal and Oro-pharyngeal swab |
1,480× |
4 |
EPI_ISL_435283 |
JKT-EIJK2444 |
Indonesia (Jakarta) |
Eijkman Institute for Molecular Biology, Ministry of Research and Technology/National Agency for Research and Innovation of the Republic of Indonesia |
Homo sapiens |
Nasopharyngeal swab |
8,082× |
5 |
EPI_ISL_437187 |
EJ-ITD853Sp |
Indonesia (Surabaya) |
Institute of Tropical Disease, Universitas Airlangga |
Homo sapiens |
Sputum |
764× |
6 |
EPI_ISL_437188 |
EJ-ITD3590NT |
Indonesia (Surabaya) |
Institute of Tropical Disease, Universitas Airlangga |
Homo sapiens |
Nasopharyngeal and Oro-pharyngeal swab |
96× |
7 |
EPI_ISL_437189 |
JKT-EIJK01 |
Indonesia (Jakarta) |
Eijkman Institute for Molecular Biology, Ministry of Research and Technology/National Agency for Research and Innovation of the Republic of Indonesia |
Homo sapiens |
Nasopharyngeal and Oro-pharyngeal swab |
2,256× |
8 |
EPI_ISL_437190 |
JKT-EIJK02 |
Indonesia (Jakarta) |
Eijkman Institute for Molecular Biology, Ministry of Research and Technology/National Agency for Research and Innovation of the Republic of Indonesia |
Homo sapiens |
Nasopharyngeal swab |
5,297× |
9 |
EPI_ISL_437191 |
JKT-EIJK03 |
Indonesia (Jakarta) |
Eijkman Institute for Molecular Biology, Ministry of Research and Technology/National Agency for Research and Innovation of the Republic of Indonesia |
Homo sapiens |
Nasopharyngeal swab |
2,112× |
10 |
EPI_ISL_437192 |
JKT-EIJK04 |
Indonesia (Jakarta) |
Eijkman Institute for Molecular Biology, Ministry of Research and Technology/National Agency for Research and Innovation of the Republic of Indonesia |
Homo sapiens |
Nasopharyngeal and Oro-pharyngeal swab |
5,759× |
Genetic Composition Analysis
We analyzed the genetic composition of SARS-CoV-2 spike glycoproteins (both nucleotide variants and amino acid changes) using Molecular Evolutionary Genetics Analysis (MEGA) X12,17. We used the Wuhan-Hu-1 isolate as a reference gene, according to Sekizuka et al. (2020)18.
Similarity Analysis
We analyzed the similarity of SARS-CoV-2 spike glycoprotein genes using the LALIGN web server (The SIB Swiss Institute of Bioinformatics, Switzerland) with an E-value threshold of 10.0. The LALIGN program is based on an algorithm first described by Huang and Miller19.
Coronaviruses infect both animals and humans and lead to various illnesses, including neurological, enteric, and respiratory diseases. There are four distinct genera of coronaviruses: Alphacoronavirus, Betacoronavirus, Gammacoronavirus, and Deltacoronavirus8. SARS-CoV, MERS-CoV, and SARS-CoV-2 are three highly pathogenic coronaviruses, capable of infecting humans, that emerged in 2002, 2012, and 2019, respectively3-4. In this study, the genetic compositions and sequence similarities of nine Indonesian SARS-CoV-2 spike glycoprotein genes were determined using sequences obtained from GenBank and the GISAID EpiCoV database (Table 2-4). Many researchers worldwide have previously reported mutations in the SARS-CoV-2 genome10,20-21, however, there has not previously been a study of the SARS-CoV-2 genome from Indonesian isolates.
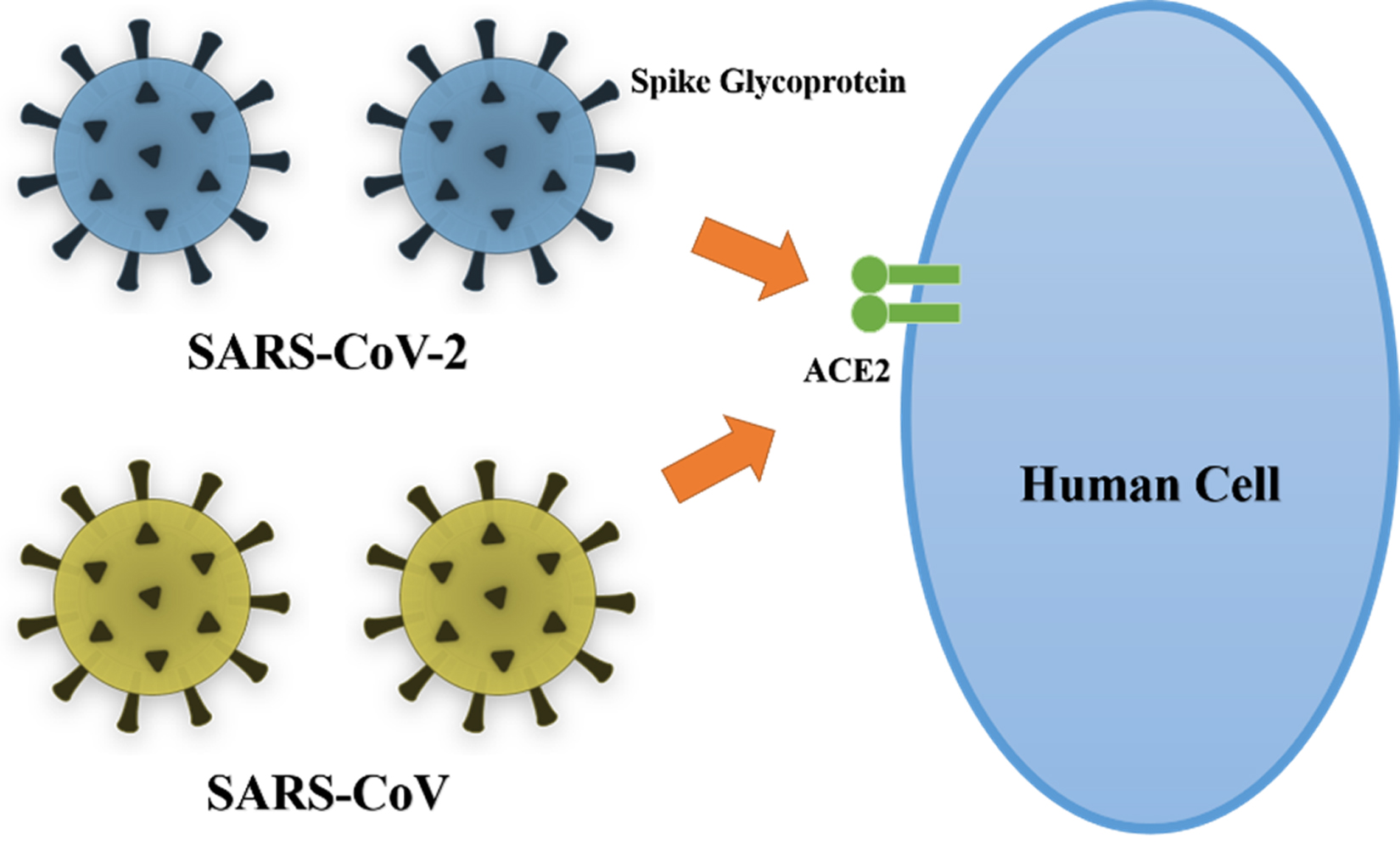
Fig. 1. Schematic diagram of SARS-COV and SARS-CoV-2 binding with high affinity to ACE2, an essential step in viral entry into host cells.
Table (2):
Nucleotide mutation sites in the SARS-CoV-2 spike glycoprotein.
| No | Virus Name | Nucleotide Position | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 224 | 347 | 414 | 1715 | 1841 | 1864 | 2031 | 2464 | ||
| 1 | Wuhan-Hu-1 (Reference) | C | C | T | C | A | G | G | C |
| 2 | JKT-EIJK0141 | C | C | T | C | A | G | G | C |
| 3 | JKT-EIJK0317 | C | C | T | C | A | G | G | C |
| 4 | JKT-EIJK2444 | T | C | T | C | A | G | G | C |
| 5 | EJ-ITD853Sp | C | C | T | C | A | G | G | C |
| 6 | EJ-ITD3590NT | C | G | T | C | G | G | T | C |
| 7 | JKT-EIJK01 | C | C | C | C | A | T | G | C |
| 8 | JKT-EIJK02 | C | C | T | C | A | G | G | C |
| 9 | JKT-EIJK03 | C | C | T | C | A | G | G | C |
| 10 | JKT-EIJK04 | C | C | T | T | A | G | G | T |
Table (3):
Amino acid mutation sites in the SARS-CoV-2 spike glycoprotein.
| No | Virus Name | Amino Acid Position | ||||||
|---|---|---|---|---|---|---|---|---|
| 76 | 116 | 572 | 614 | 622 | 677 | 822 | ||
| 1 | Wuhan-Hu-1 (Reference) | T | S | T | D | V | Q | L |
| 2 | JKT-EIJK0141 | T | S | T | D | V | Q | L |
| 3 | JKT-EIJK0317 | T | S | T | D | V | Q | L |
| 4 | JKT-EIJK2444 | I | S | T | D | V | Q | L |
| 5 | EJ-ITD853Sp | T | S | T | D | V | Q | L |
| 6 | EJ-ITD3590NT | T | C | T | G | V | H | L |
| 7 | JKT-EIJK01 | T | S | T | D | F | Q | L |
| 8 | JKT-EIJK02 | T | S | T | D | V | Q | L |
| 9 | JKT-EIJK03 | T | S | T | D | V | Q | L |
| 10 | JKT-EIJK04 | T | S | I | D | V | Q | F |
Table (4):
Sequence similarity of SARS-CoV-2 spike glycoprotein genes, determined using the LALIGN web server.
| Virus Name | Similarity | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Wuhan-Hu-1 (Reference) | JKT-EIJK0141 | JKT-EIJK0317 | JKT-EIJK2444 | EJ-ITD853Sp | EJ-ITD3590NT | JKT-EIJK01 | JKT-EIJK02 | JKT-EIJK03 | |
| JKT-EIJK0141 | 100% | ||||||||
| JKT-EIJK0317 | 100% | 100% | |||||||
| JKT-EIJK2444 | 100% | 100% | 100% | ||||||
| EJ-ITD853Sp | 100% | 100% | 100% | 100% | |||||
| EJ-ITD3590NT | 99.9% | 99.9% | 99.9% | 99.9% | 99.9% | ||||
| JKT-EIJK01 | 99.9% | 99.9% | 99.9% | 99.9% | 99.9% | 99.9% | |||
| JKT-EIJK02 | 100% | 100% | 100% | 100% | 100% | 99.9% | 99.9% | ||
| JKT-EIJK03 | 100% | 100% | 100% | 100% | 100% | 99.9% | 99.9% | 100% | |
| JKT-EIJK04 | 99.9% | 99.9% | 99.9% | 99.9% | 99.9% | 99.9% | 99.9% | 99.9% | 99.9% |
The coronavirus spike glycoprotein mediates membrane fusion and viral entry into host cells and is therefore the primary target for many neutralizing antibodies (Fig. 1). The spike glycoprotein has two domains, S1 and S2, where S1 is responsible for binding the virion to ACE2 on the host cell membrane21. Several antiviral drugs and vaccines have been developed which target the spike glycoprotein. Du et al. (2009)22 demonstrated the effectiveness of several of these antiviral therapies, including small interfering RNAs, protease inhibitors, ACE2 blockers, fusion blockers, spike glycoprotein inhibitors, neutralizing antibodies, and spike glycoprotein cleavage inhibitors in in vitro studies. In addition, a number of techniques have been used to generate vaccines using all or part of the spike glycoprotein as an antigen. These include the use of recombinant receptor binding domain protein, viral vectors, full-length S protein, recombinant spike glycoprotein, and spike protein DNA-expressing vectors. Thus, it is very important to investigate the genetic composition and sequence similarity of this protein.
Interestingly, despite reporting several mutation sites in this study, we demonstrate that there is no significant change in the genetic composition of SARS-CoV-2 spike glycoprotein genes. Nucleotide variants in SARS-CoV-2 spike glycoprotein genes described in this study include JKT-EIJK2444 (224C>T), EJ-ITD3590NT (347C>G; 1841A>G; 2031G>T), JKT-EIJK01 (414T>C; 1864G>T), and JKT-EIJK04 (1715C>T; 2464C>T) (Table 2). In addition, we also analyzed amino acid changes in the SARS-CoV-2 spike glycoprotein including JKT-EIJK2444 (T76I), EJ-ITD3590NT (S116C; D614G; Q677H), JKT-EIJK01 (V622F), and JKT-EIJK04 (T572I; L822F) (Table 3). Finally, the interval score of similarity between each of the SARS-CoV-2 spike glycoproteins was between 99.9% and 100% (Table 4).
Previous studies investigating gene variability in SARS-CoV-2 samples include the work of Sekizuka et al. (2020), who performed whole-genome sequencing of SARS-CoV-2, directly from PCR-positive clinical specimens18. This was conducted in order to generate a haplotype network analysis of the Diamond Princess cruise ship outbreak, using the Wuhan-Hu-1 sequence as a reference. Additionally, Castillo et al. (2020) reported a phylogenetic analysis of the first four SARS-CoV-2 cases in Chile, analyzing nucleotide variants, amino acid changes, and sequence similarity23. While our study complements these previous works, several shortcomings remain, including the relatively small number of isolates studied, the methodology used for whole-genome sequencing, and the quality coverage of SARS-CoV-2 genomes isolated from Indonesia.
As new information on SARS-CoV-2 is published daily, new concepts and frameworks must constantly be adopted. Currently, the GISAID EpiCoV database and Tang et al. (2020) have established three subtypes of SARS-CoV-2 based on nucleotide variants that produce amino acid changes: S, G, and V24. The mutation rate of viruses is considerably higher than most other biological entities, including prokaryotes and eukaryotes. This is especially true of RNA-based viruses such as SARS-CoV-2, Ebola and dengue, due to hydroxyl groups in RNA that act as catalytic sites for mutation. This advanced mutation rate, leads to enhanced virulence and a higher capacity for adaptive evolution25-26. While Tang et al. (2020) have suggested that SARS-CoV-2 exhibits the characteristic high mutation rate of an RNA virus24, in fact, the mutation rate of SARS-CoV-2 and other coronaviruses might be slightly lower than other RNA viruses because of its genome-encoded exonuclease. Regardless, its high mutation rate increases the potential of this zoonotic viral pathogen to adapt to efficient transmission from human to human and potentially allows it to become more virulent.
The genomic characteristics of SARS-CoV-2 are significantly different than either SARS-CoV or MERS-CoV27. A previous study reported that the homology of SARS-CoV-2 with the bat coronavirus isolate, RaTG13, was 96%28. Interestingly, another study reported that the homology of SARS-CoV-2 with a pangolin coronavirus was 99%1. From these results, it could be suggested that pangolins act as an intermediate host between bats and humans.
This study of genomic variants of SARS-CoV-2 isolated from Indonesia is crucial for future investigations into the pathogenesis, prevention, and treatment of SARS-CoV-2. Development of this genomic data is vital work that will facilitate vaccine design, epidemiological investigations, viral detection, functional analysis, and evaluation of treatment options27.
Outbreaks of SARS-CoV-2 have led to a state of medical and economic emergency worldwide. Therefore, understanding the characteristics of the SARS-CoV-2 genome and developing systems to monitor SARS-CoV-2 during the pandemic are critical steps for controlling this disease. The identification of genotypes connected to specific geographic and temporal infectious clusters suggests that genomic data can be used to track and monitor the transmission of SARS-CoV-2. Therefore, the rapid discovery of genetic variants of SARS-CoV-2 is necessary for a streamlined response to the COVID-19 outbreak. Similarly, identifying specific SARS-CoV-2 variants and connecting them using a molecular epidemiology approach would allow researchers to determine the origin of a specific variant and monitor its transmission. This could be an important tool in controlling the outbreak21.
In summary, there was no significant difference between the SARS-CoV-2 spike glycoprotein gene sequences found in Indonesia and the Wuhan-Hu-1 isolate from China. However, this was only a preliminary study and we recommend expanding molecular epidemiology and surveillance programs to monitor COVID-19 in Indonesia.
ACKNOWLEDGMENTS
This study was supported by the Directorate General of Higher Education, Ministry of Education and Culture of the Republic of Indonesia; the Institute of Research and Community Empowerment (LPPM) of the Indonesia International Institute for Life Sciences (I3L); and Generasi Biologi Indonesia (GENBINESIA) Foundation, Indonesia. We thank Editage for editing the manuscript.
CONFLICT OF INTEREST
The authors declares that there is no conflict of interest.
AUTHORS’ CONTRIBUTION
All listed authors made a substantial, direct, and intellectual contribution to the work and approved it for publication.
FUNDING
PMDSU Scholarship Batch III from the Directorate General of Higher Education, Ministry of Education and Culture of the Republic of Indonesia.
ETHICS STATEMENT
This article does not contain any experiments using human participants or animals performed by any of the authors.
AVAILABILITY OF DATA
Not applicable.
- Lam TT, Shum MH, Zhu HC, et al. Identifying SARS-CoV-2 related coronaviruses in Malayan pangolins. Nature. 2020.
Crossref - Gorbalenya AE, Baker SC, Baric RS, et al. The species severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol. 2020;5:536-544.
Crossref - Andersen KG, Rambaut A, Lipkin WI, Holmes EC, Garry RF. The proximal origin of SARS-CoV-2. Nat Med. 2020;26:450-452.
Crossref - Kharisma VD, Ansori ANM. Construction of epitope-based peptide vaccine against SARS-CoV-2: Immunoinformatics study. J Pure Appl Microbiol. 2020;14:6248.
- Lai CC, Shih TP, Ko WC, Tang HJ, Hsueh PR. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease-2019 (COVID-19): The epidemic and the challenges. Int J Antimicrob Agents. 2020;55:105924.
Crossref - Dong E, Du H, Gardner L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect Dis. 2020;S1473-3099(20): 30120-30121.
Crossref - Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell, 2020;181:281-292.e6.
Crossref - Ou X, Liu Y, Lei X, et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat Commun. 2020;11:1620.
Crossref - Shereen MA, Khan S, Kazmi A, Bashir N, Siddique R. COVID-19 infection: Origin, transmission, and characteristics of human coronaviruses. J Adv Res. 2020;24:91-98.
Crossref - Phan T. Genetic diversity and evolution of SARS-CoV-2. Infect Genet Evol. 2020;81:104260.
Crossref - Ansori ANM, Sucipto TH, Deka PT, et al. Differences of universal and multiplex primer for detection of Dengue virus from patients suspected Dengue Hemorrhagic Fever (DHF) in Surabaya. Indonesian J Trop Infect Dis. 2015;5:147-151.
Crossref - Ansori ANM, Kharisma VD. Characterization of Newcastle disease virus in Southeast Asia and East Asia: Fusion protein gene. Eksakta. 2020;1:20-28.
- Callaway E. The race for coronavirus vaccines: A graphical guide. Nature. 2020;580:576-577.
Crossref - Shang W, Yang Y, Rao Y, Rao X. The outbreak of SARS-CoV-2 pneumonia calls for viral vaccines. NPJ Vaccines. 2020;5:18.
Crossref - Al-Tawfiq JA, Al-Homoud AH, Memish ZA. Remdesivir as a possible therapeutic option for the COVID-19. Travel Med Infect Dis. 2020;34:101615.
Crossref - Wang M, Cao R, Zhang L, et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020;30:269-271.
Crossref - Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol Biol Evol. 2018;35:1547-1549.
Crossref - Sekizuka T, Itokawa K, Kageyama T, et al. Haplotype networks of SARS-CoV-2 infections in the Diamond Princess cruise ship outbreak. medRxiv, 2020.
Crossref - Joob B, Wiwanitkit V. Genetic variant severe acute respiratory syndrome coronavirus 2 isolates in Thailand. J Pure Appl Microbiol. 2020;14:6314.
Crossref - Benvenuto D, Angeletti S, Giovanetti M, et al.. Evolutionary analysis of SARS-CoV-2: How mutation of Non-Structural Protein 6 (NSP6) could affect viral autophagy. J Infect. 2020;S0163-4453(20):30186-9.
Crossref - Yin C. Genotyping coronavirus SARS-CoV-2: methods and implications. Genomics. 2020;S0888-7543(20):30318-9.
Crossref - Du L, He Y, Zhou Y, Liu S, Zheng B-J, Jiang S. The spike protein of SARS-CoV–a target for vaccine and therapeutic development. Nat Rev Microbiol, 2009; 7: 226e36.
Crossref - Castillo AE, Parra B, Tapia P, Acevedo A, Lagos J, Andrade W, Arata L, Leal G, Barra G, et al. Phylogenetic analysis of the first four SARS-CoV-2 cases in Chile. J Med Virol. 2020. [Epub ahead of print]
Crossref - Tang X, Wu C, Li X, et al. On the origin and continuing evolution of SARS-CoV-2. Natl Sci Rev, 2020; nwaa036.
- Duffy S. Why are RNA virus mutation rates so damn high? PLoS Biol. 2018;16:e3000003.
Crossref - Eyer L, Nencka R, de Clercq E, Seley-Radtke K, Ruzek D. Nucleoside analogs as a rich source of antiviral agents active against arthropod-borne flaviviruses. Antivir Chem Chemother. 2018;26:2040206618761299.
Crossref - Wang C, Liu Z, Chen Z, Huang X, Xu M, He T, Zhang Z. The establishment of reference sequence for SARS-CoV-2 and variation analysis. J Med Virol. 2020.
Crossref - Zhou P, Yang XL, Wang XG, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270-273.
Crossref
© The Author(s) 2020. Open Access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License which permits unrestricted use, sharing, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.