


ISSN: 0973-7510
E-ISSN: 2581-690X
Leptospirosis is a neglected zoonosis caused by a pathogenic spirochete, Leptospira interrogans. The mode of infection in humans is through an abrasion in human skin or the conjunctiva and mucous membrane. Infected patients usually show different symptoms resembling bacterial or viral infections such as the flu. Hence, diagnosing leptospirosis in the early stage is complex, and can be easily confused with other infections. A strategical pathway was developed to analyze the hypothetical proteins in L. interrogans and unveil their potential as diagnostic markers. Subcellular localization tools such as PSORTb, CELLO, SOSUI-GramN, and ProtCompB were used to segregate the outer membrane and surface proteins from the overall pool of hypothetical proteins. The shortlisted proteins were checked for their virulency, and antigenicity through tools such as VirulentPred, and VaxiJen, respectively. Proteins with the highest scores were fed into ElliPro which predicted both linear and discontinuous epitopes in each protein. Proteins with many epitopes were further analyzed with BepiPred 3.0, which provided the epitope probability for each protein’s amino acid. Epitope probability of the potential proteins was compared with the standard diagnostic marker, LipL32. The comparison revealed that a protein (UniProt ID D4YW28) has better immunogenic potential than the gold standard marker, LipL32. In conclusion, this protein can be used as a diagnostic marker for the detection of leptospirosis and it will also serve as a better vaccine candidate.
Leptospirosis, Diagnosis, Epitope, Localization, Antigenicity, Hypothetical Proteins
Leptospirosis is now a major concern being a tropical zoonotic disease, posing a severe threat to mankind who survive natural disasters such as floods and hurricanes which leads to an outbreak of the disease.1 A total of 21 species of Leptospira, the notorious spirochete, have been identified of which 240 serovars have been reported, where only a few are disease-causing pathogenic serovar that comes under the species of L. interrogans.2 The World Health Organization depicts around 24 serogroups of Leptospira, classified based on their similar antigenicity. Transmission of the disease to humans is both from direct contact with infected animals in the cases of people working in slaughterhouses, and animal farms or indirectly by getting exposed to contaminated water or soil.3 The reservoir hosts of L. interrogans are very diverse, as they tend to be carried by rodents, canines, and other livestock. The World Health Organization states that rodents carry L. interrogans throughout their lifespan without any clinical manifestations, resulting in the constant shedding of leptospires in their urine, making humans more vulnerable to the disease.
The clinical manifestations of leptospirosis in humans are found to be varying, ranging from acute febrile illness to chronic stage of multiple organ failure invading the lungs, liver, and kidney.4 Patients with leptospirosis show biphasic symptoms, starting with the acute phase, where patients present high fever, and proceeding with the immune phase which involves antibody production in the host.5 Pulmonary manifestations of leptospirosis lead to a haemorrhagic syndrome in the lungs mimicking several other viral fevers that were used in bioterrorism.6 Severe complications of leptospirosis include hepatic failure causing Weil’s disease with clinical presentations of a cholestatic pattern of the liver.7 Early treatment of leptospirosis is of dire need given the fatal neurological condition such as meningitis that requires testing of cerebrospinal fluid through a lumbar puncture in patients.8
Leptospires can be visualized directly under Dark-field or phase-contrast microscopy as they lack proper staining techniques.9 Microscopic agglutination test (MAT-gold standard) and IgM ELISA are the traditional antibody-based methods of diagnosis for clinical interventions of infected people.3 MAT fails to diagnose leptospirosis in the acute phase of infections which led to the development of rapid screening tests such as macro-agglutination test, indirect hemagglutination assay, microcapsule agglutination test, latex agglutination test, enzyme-linked immunosorbent assay (ELISA), dot enzyme immunoassay (dipstick), lateral flow assay (LFA), and immunofluorescence assay.1 The lateral flow assay-based diagnosis is biased in their accuracy, hence a secondary confirmation through MAT and nucleic acid amplification tests (NAAT) has always been recommended after rapid tests. Unlike MAT, the NAATs have high sensitivity in predicting leptospirosis in the acute phase of infection.10 The existing NAATs include polymerase chain reaction (PCR), and Loop-Mediated Isothermal Amplification (LAMP) targeting housekeeping genes, and species-specific genes. lipL32 is the most commonly targeted gene among all the other biomarker genes such as loa22, ompL1, ligB, etc., owing to its presence in the pathogenic L. interrogans.11 Experimental studies have been carried out to analyze the known outer membrane proteins, OmpL1, surface-exposed lipoproteins, LigA and LigB, secretary proteins, sphingomyelinase C (SphA) and hemolysin (SphH) and lipoproteins, LipL32, LipL41, LipL36, and LipL21, on account of their host-pathogen interactions and pathogenicity.12 The virulence of pathogenic L. interrogans was proven to be conferred by the protein Loa22 which has a 73% similar ortholog in saprophytic Leptospira biflexa. This proves that the protein is essential for the environmental existence of the genus.13 Though LipL32 accounts for 75% of the outer membrane proteins of L. interrogans, it does not play a vital role in the virulence or survival of the organism which has been proved by lipL32 gene mutant studies.14 Despite being exclusive to pathogenic species, the role of LipL32 protein in the pathogenesis of L. interrogans is unclear which makes us question its potential to be a biomarker.
One-third of the genome of every organism reflects hypothetical proteins that have been left uncharacterized.15 Annotation and analysis of hypothetical proteins are essential as they reveal many useful proteins involved in host-pathogen interactions that could serve as efficient diagnostic/vaccine candidates.16 Many in silico approaches have been carried out in search of potential vaccine candidates in the hypothetical, and putative protein pool of L. interrogans which fell under the extracellular and outer membrane proteins category.17
In this study, a stepwise approach is established by adopting prediction algorithms such as subcellular localization, virulency and antigenicity analysis, epitope profile, and protein-protein interaction studies, to find potential biomarkers among the hypothetical proteins of L. interrogans serovar Lai. Analysing all the hypothetical proteins is very cumbersome which calls for a simplified protocol with proper control over the flow to end up with a significant protein. Predicting the subcellular localization of the hypothetical proteins is the first step of the strategy to pick the outer membrane and extracellular proteins which can serve as better diagnostic candidates. The generation of epitope and antigenicity profiles for the proteins is the key step in this study, which looks for the probable interaction of the query protein with antibodies with the help of epitope prediction algorithms. B lymphocyte cells (B cells), a type of blood cell involved in the humoral immune system, get activated by antigen binding and differentiate into antibody-secreting plasma cells.18 Epitopes are the regions of antigenic proteins that are recognized by the specific antibodies secreted by activated B cells.19 Epitopes are mostly discontinuous, i.e., a cluster of amino acids (not necessarily adjacent in sequence) brought in close proximity by their 3D conformation of the protein.20 Since B cell epitopes have a crucial role in immune response, recruiting them as diagnostic markers became necessary. This study presumes that the proteins can be enlisted under potential biomarkers upon satisfying the requirements of having a higher number of discontinuous epitopes. To validate the significant biomarkers, their antigenicity and epitope profiles were compared with the LipL32 protein.
Sequence retrieval
The genome of L. interrogans serovar Lai 56601 (KEGG entry-T00098) from the KEGG Genome Database (https://www.genome.jp/kegg/genome/) was chosen for this study. A total of 3754 genes were linked to the genome entry, comprising 1363 hypothetical proteins from both the chromosomes of L. interrogans which constitutes around 36% of the genome. The hypothetical protein (HP) sequences were retrieved from UniProt KB (https://www.uniprot.org/) using the accession number provided in the KEGG genes database.
Sub-cellular localization
In the quest for cell surface biomarkers for the detection of L. interrogans, localization of the marker protein becomes an important factor. Surface outer membrane proteins are of greater concern in order to categorize them as potential biomarkers. Hypothetical proteins of L. interrogans serovar Lai that belonged to the outer membrane and extracellular protein family were shortlisted for further study. Four subcellular localization (SCL) tools were adopted for the analysis: PSORTb,21 CELLO,22 SOSUI-GramN,23 ProtCompB.24 TMHMM25 was used to predict the number of transmembrane helices in a protein.
Protparam-Expasy and InterProScan analysis
ProtParam26 computes various physicochemical properties that can be deduced from a protein sequence. The ProtParam provides information on the molecular weight, theoretical pI, amino acid composition, atomic composition, extinction coefficient, estimated half-life, instability index, aliphatic index, and grand average of hydropathicity (GRAVY). The hypothetical proteins belonging to the outer membrane and extracellular category were analyzed with ProtParam tool. The functional analysis of proteins was done by InterProScan (https://www.ebi.ac.uk/interpro/search/sequence/) which helps in obtaining information on homology, ontology, domain, and family of the proteins. Gene ontology (GO) information was provided in three categories such as cellular component, molecular function, and biological process.
Protein-Protein interactions using STRING database
Functional analysis of genes requires finding three genomic context associations which include conserved genomic neighborhood, gene fusion events, and co-occurrence of genes across various genomes.27 STRING database has been widely used for predicting protein-protein interactions where it uses the COG database to acquire all the orthology information for the given set of genes.28 It also uses protein mode, in which the Smith-Waterman algorithm was used to select the potential orthologs of the chosen pair of proteins in other genomes.29 The predicted association of a gene to a pathway/function was scored based on a reference dataset of true associations. STRING version 11.530 was used for constructing a protein-protein interaction (PPI) network between the shortlisted outer membrane proteins.
Virulence prediction and antigenicity prediction
The outer membrane and extracellular proteins selected from the HP pool were fed into the VirulentPred tool to segregate the virulent proteins. The Virulent Pred31 server uses the SVM classifier’s bilayer cascade to provide optimal results through double-layer verification. Proteins with a higher score of above 1 were considered virulent, and a score of less than zero (negative) were considered avirulent.
VaxiJen32 uses Perl as its programming language, with an HTML interface to predict the antigens. The shortlisted virulent proteins from the HP pool of L. interrogans were checked for their antigenic potential, and proteins with a higher VaxiJen score of above 0.7 were considered for further analysis.
Epitope prediction by ElliPro and BepiPred 3.0
Virulent hypothetical proteins having high antigenic potential were further screened for the presence of epitope regions. ElliPro was used to predict the linear and discontinuous epitopes in the significant proteins that had high VaxiJen scores. ElliPro33 performed structure-based epitope prediction; hence, 3D structures of proteins were uploaded as PDB files. ElliPro listed the number of continuous and discontinuous epitopes from the protein and the position of the residues involved. The Server also provides a visualization option to view the 3D structure of the epitope region using Jmol viewer.
BepiPred 3.0 server34 was used to predict the B cell epitopes in the hypothetical proteins that had more than 7 discontinuous epitopes. BepiPred-3 provides epitope probability for each amino acid in a protein sequence, which was then plotted as a graph having 0.1512 as a threshold epitope score. B cell epitopes present in LipL32 were also analyzed in order to compare with the shortlisted hypothetical proteins.
Sub-cellular localization
Nucleic acid amplification-based leptospirosis diagnostics methods depend on a marker genes that are involved in the pathogenicity. Using bioinformatic approaches, an extensive in silico analysis of the putative outer membrane and extracellular proteins of L. interrogans serovar Lai and Copenhageni genome has been carried out in a earlier study,17 to annotate the uncharacterized hypothetical proteins. In silico and microarray-based processes have aided in predicting potential vaccine candidates against leptospirosis, which further seeks experimental evidence for vaccine studies.35 Novel vaccine candidates were searched in L. interrogans through whole genome analysis where the study mainly focused on the hypothetical proteins with lipo-box motifs, which was predicted through PSORT and SignalP bioinformatic tools.36
In the present study, we analyzed the hypothetical proteins of L. interrogans serovar Lai in search of potential marker proteins. A total of 1363 hypothetical proteins were analyzed with localization tools (PSORTb, CELLO, SOSUI-GramN, and ProtCompB). The performance of the SCL tools was generally validated by running experimentally known proteins as a training set whose biological functions had already been identified. The accuracy of the subcellular localization tools is generally predicted through Matthew’s correlation coefficient (MCC) which is a performance predictive metric that holds a value of one for a perfect prediction and zero for any false assignment.37 The overall performance accuracy is a ratio between the number of correct predictions and the total number of sequences. The PSORTb 3.0 server (updated version) used in this study possesses a higher precision of 97.3% for SCL prediction in Gram-negative bacteria, whereas CELLO has a precision of 87.5%.21 The ProtCompB has a higher recall rate of 79% with a precision of 83%.17 The performance of SOSUI-GramN was evaluated using test data and its precision was found to be 92.3% and 89.4% for extracellular and outer membrane proteins respectively.23 The hypothetical proteins that lack annotations in the databases were handled in the study, so it was impossible to calculate the MCC or precision percentage without the knowledge of true positives and false negatives of the prediction methodology. Hence the SCL tools were chosen based on their established precision and recall rate. MetaLocGramN is an SCL tool that uses features from four predictors (PSORTb 3.0, PSLPred, CELLO, and SOSUI-GramN) for accurate prediction with their combined strengths38 proving the potential characteristics of the SCL tools used in this study. The SCL tools used in this study come under the category of homology-based prediction tools that use annotated and experimentally identified proteins as templates with the aid of Support Vector Machines (SVM) and deep neural networks (CNN). One of the limitations of SCL while predicting the location of an unannotated protein is that they do not have a promising template to depend on.39
The HP sequences are uploaded in the localization tools and the predictions are given in Supplementary Material 1, Table S1. The consensus and non-consensus vote-based selection of the proteins have been followed in an earlier study where three SCL tools were tested with experimentally known proteins wherein the precision, and recall rates of their performances were predicted.17 The present study adopted a similar method since SCL prediction was the first step in selecting potential proteins from the unknown pool. Based on the majority votes i.e. if 3 out of 4 SCL tools predicted a protein as outer membrane or extracellular, it is considered a consensus vote and the protein was shortlisted. Further, the proteins were screened with TMHMM, and those with 0/1 transmembrane helix were chosen for further analysis. Many surface adhesins such as LigA, and LigB are located on the surface of the L. interrogans which are considered potential biomarkers for diagnosis due to their pathogenicity.40 The study aims to obtain such hypothetical proteins that come in the close proximity to the host proteins and have antigenic effects. The proteins must not be completely spanned in the membrane to be on the surface. Hence the proteins that qualify as outer membrane/extracellular proteins with 1/0 helices were preferred for further analysis.
Proteins, a count of 129 that fell under the desired category, were taken further through the epitope prediction pathway. The chromosome-wise distribution of the shortlisted proteins is given in Figure 1. The findings of the subcellular localization listed around 129 proteins categorized under the outer membrane and extracellular proteins. The thumb rule of having less than 2 transmembrane helices narrowed the analysis down to a smaller number of proteins by having a stringent control. This particular control was necessary for our study, as the search was executed to identify the diagnostic marker present in the outer surface of L. interrogans.
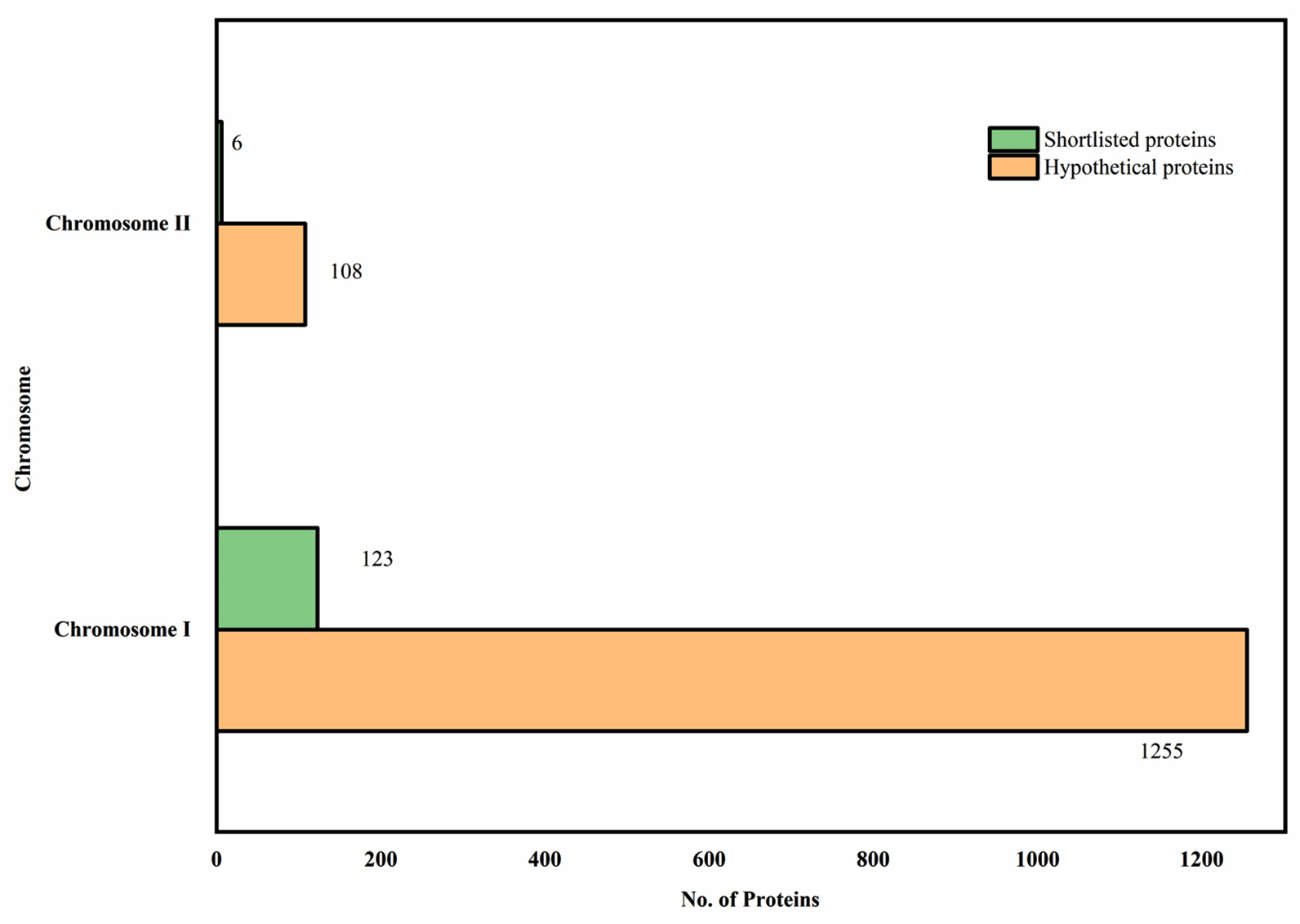
Figure 1. Outer membrane and Extracellular proteins in the hypothetical protein pool
Subcellular localization of the hypothetical proteins, providing results on the shortlisted (Green) outer membrane and extracellular proteins with 0/1 transmembrane helices in both the chromosomes of Leptospira interrogans serovar Lai
Functional analysis
ProtParam-Expasy analysis was done for the shortlisted 129 proteins to have an overview of their physicochemical parameters, and the results are listed in Supplementary Material 3, Table S1. Pre-identified GO information was retrieved for a few hypothetical proteins out of 129 from InterProScan. GO terms were pre-described in the InterProScan for 30 HPs which provided information on their cellular component. Similarly, GO terms describing the molecular functions were fetched for 31 HPs. The categorical distribution of pre-predicted GO terms of the shortlisted proteins based on their molecular function and cellular component was plotted in a bubble chart given in Figure 2. The protein-protein interactions for the 129 proteins were studied with the help of the STRING database and the results from the database were given in Figure 3.
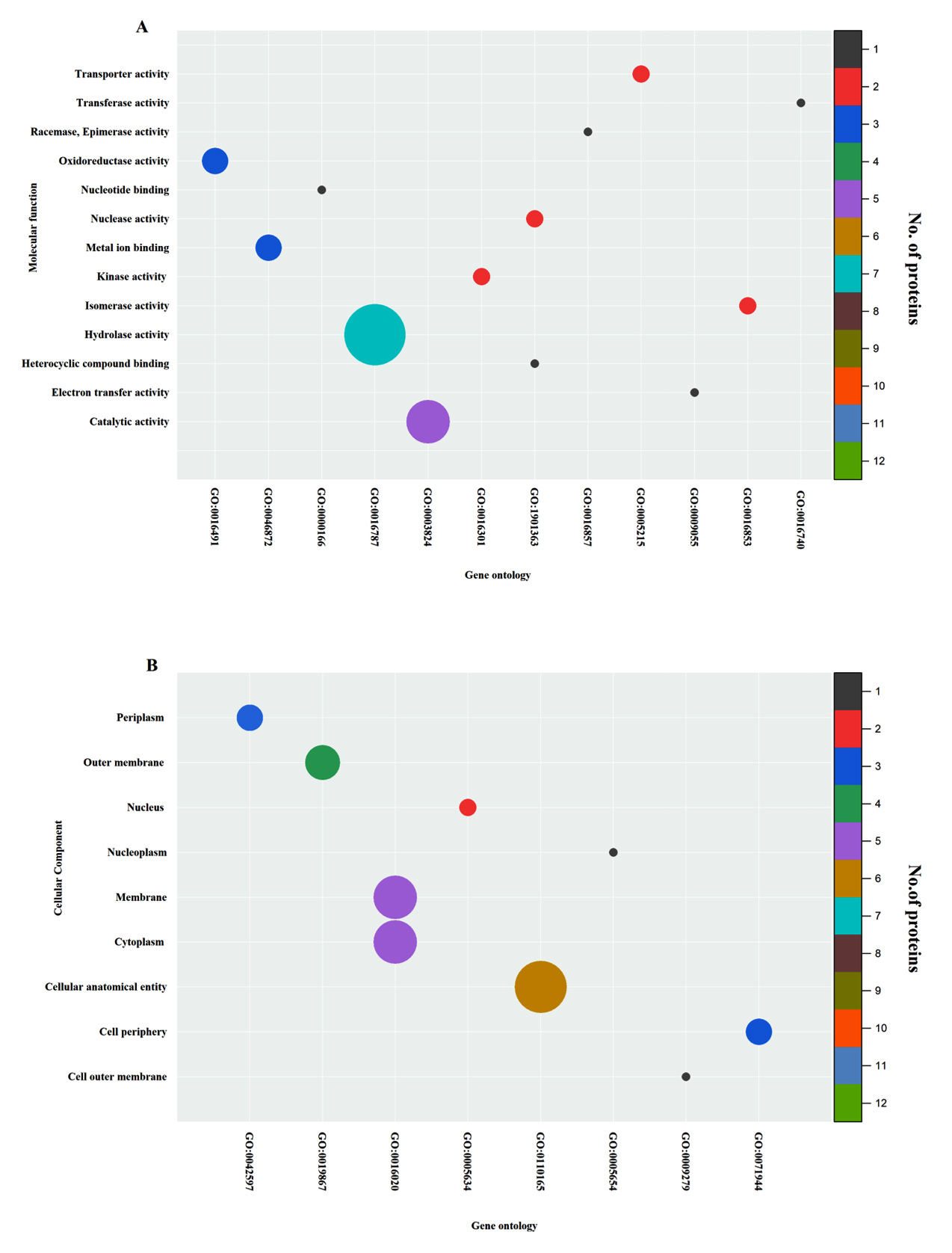
Figure 2. Bubble plots representing the Gene Ontology of shortlisted hypothetical proteins
(a) Gene Ontology of the shortlisted hypothetical proteins categorized under GO terms of Molecular function (axes denote the GO terms and name of the function in X and Y respectively, where as the color and size of the bubble represent the protein count fell under particular function) Similar procedure was used to represent the succeeding bubble plots (b) Gene Ontology of the shortlisted hypothetical proteins categorized under GO terms of Cellular component
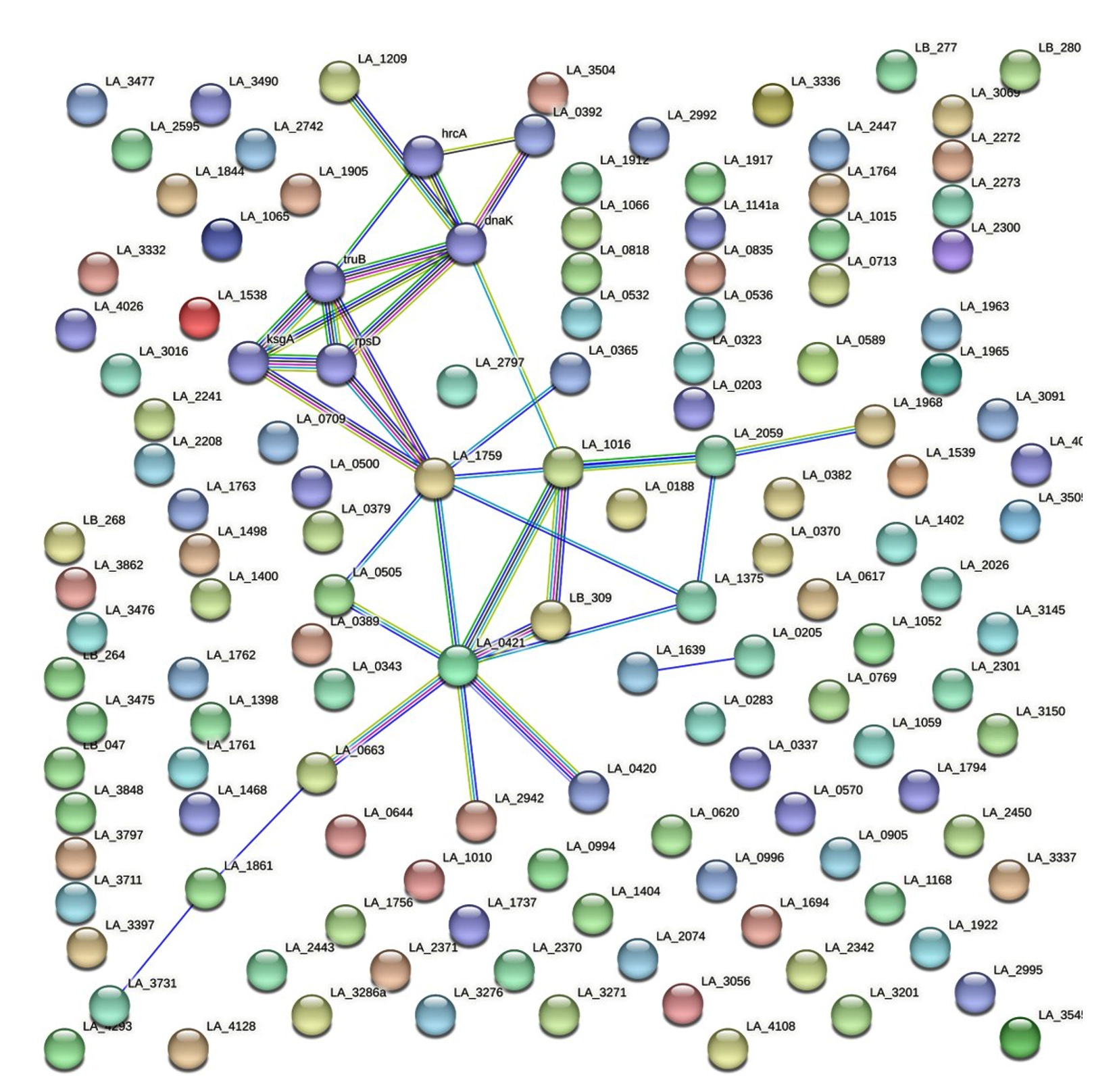
Figure 3. Protein-Protein interactions network
PPI network of the shortlisted 129 hypothetical proteins obtained from STRING database
Virulence prediction and antigenicity prediction
Virulent proteins are involved in host-pathogen interactions to confer pathogenicity, but only a few proteins can evoke immune responses by interacting with the B cells and T cells of the human body. The virulence nature of 129 proteins was predicted through VirulentPred whose virulent scores are listed in Supplementary Material 2, Table S1. Among the 129 proteins, 80 proteins had a higher score of more than 1 and these proteins were further shortlisted for the antigenicity and epitope analysis. Proteins with a score of less than 1 were considered less virulent and were eliminated from the study. A graph is given in Figure 4, where the proteins were segregated based on their virulent score obtained from VirulentPred.
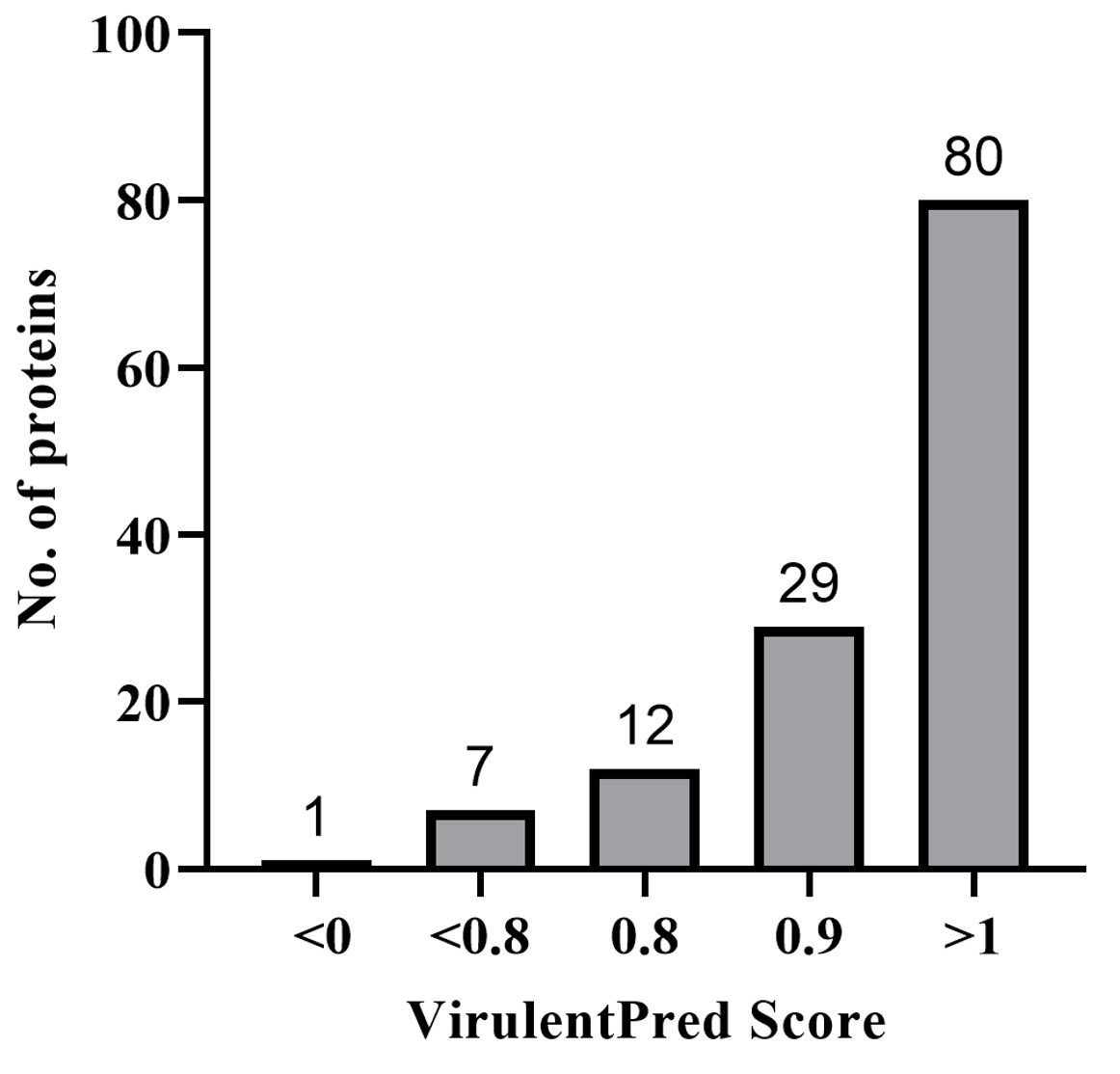
Figure 4. Virulence prediction results from VirulentPred Screening of virulence nature for 129 shortlisted hypothetical proteins by means of VirulentPred score. Proteins were grouped based on the score obtained. 80 proteins having a score of greater than 1 were considered highly virulent proteins whereas protein with a score of less than 0 was considered avirulent
The 80 virulent proteins were further screened with the VaxiJen tool for their potential behavior as antigens. The hypothetical proteins with their respective VaxiJen scores are given in Supplementary Material 2, Table S2. A total of 14 Proteins with higher VaxiJen scores, i.e. those having greater than 0.7 were selected for the epitope analysis. The antigenicity profile of the 14 proteins and LipL32, based on their scores can be found in Figure 5.
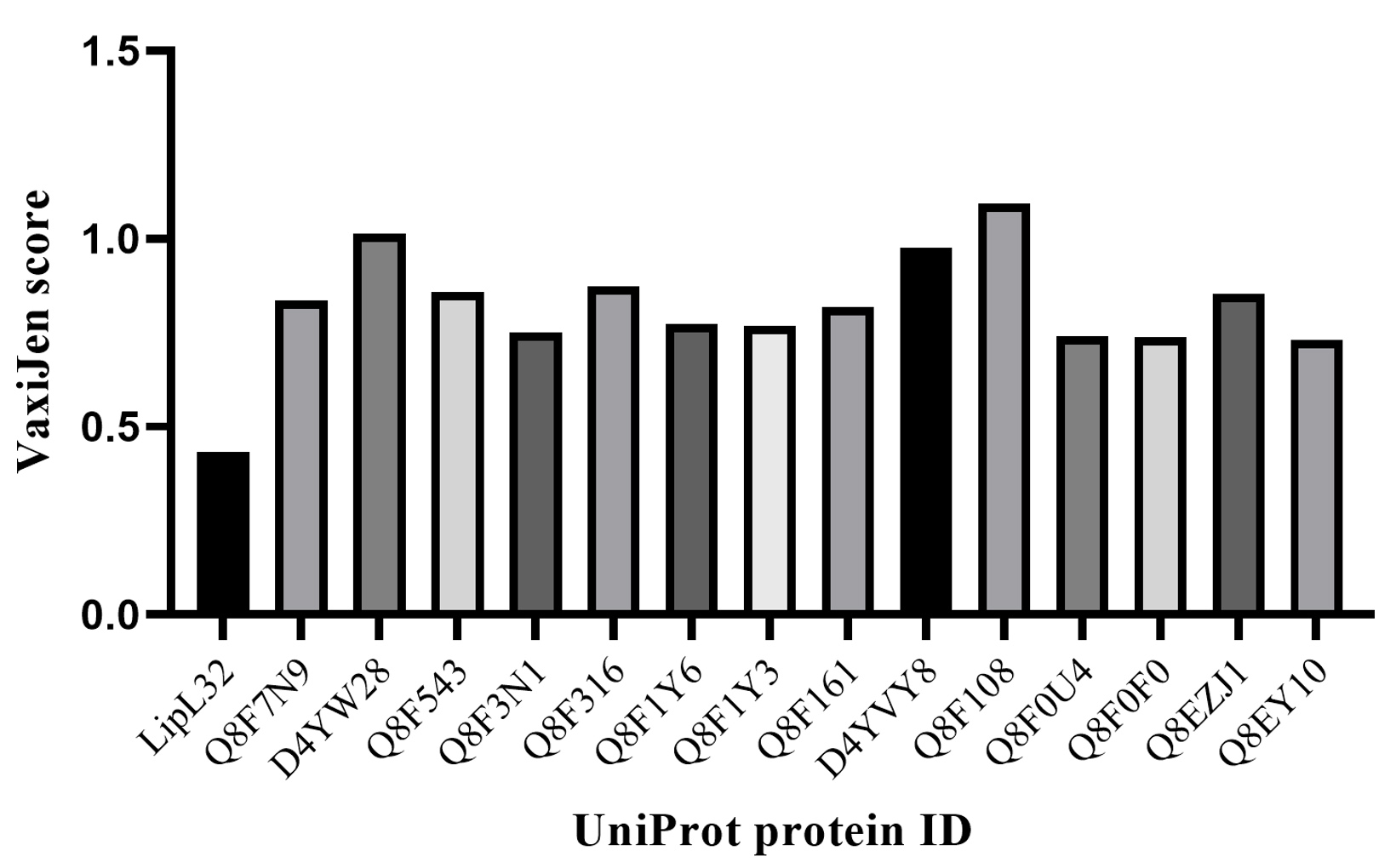
Figure 5. Potential marker proteins selection through VaxiJen
A Comparison of 14 proteins (having a high VaxiJen score above 0.7) with LipL32 is presented in the graph
Epitope prediction by ElliPro and BepiPred 3.0
This study emphasized that the predicted proteins should have more discontinuous epitopes which again trimmed the list of HP proteins. The ElliPro epitope prediction tool provided the list of linear and discontinuous epitopes for 14 significant proteins. All the predictions have been listed in Table S3, and S4 of Supplementary Material 2. A categorical graph on the number of epitopes per classification was presented in Figure 6, for the selected 14 significant proteins. It was found that among the 14 proteins, 6 were having more than 7 discontinuous epitopes. The analysis was then narrowed down to 6 proteins, namely D4YW28, Q8F3N1, Q8F1Y3, Q8F0U4, Q8EZJ1, and Q8EY10. The proteins D4YW28 and Q8EZJ1 had a count of 12 discontinuous epitopes whereas the rest of the proteins had only 8 discontinuous epitopes. The consolidated results of the six selected proteins are given in Table.
Table :
Consolidated results of selected potential biomarkers
| Subcellular Localization | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Protein ID | PSORTb | CELLO | SOSUI GramN | ProtCompB | Deep THMMM | |||||||||||
| D4YW28 | Extracellular | Extracellular and outer membrane | Inner membrane | Extracellular and outer membrane | 1 | |||||||||||
| Q8F3N1 | Unknown | Extracellular and Periplasmic | Inner membrane | Extracellular and outer membrane | 0 | |||||||||||
| Q8F1Y3 | Unknown | Extracellular and Cytoplasmic | Extracellular | Cytoplasmic | 0 | |||||||||||
| Q8F0U4 | Unknown | Outer membrane | Outer membrane | Extracellular | 0 | |||||||||||
| Q8EZJ1 | Outer membrane | Extracellular and Outer membrane, periplasmic | Inner membrane | Outer membrane | 1 | |||||||||||
| Q8EY10 | Unknown | Extracellular | Extracellular | Extracellular and periplasmic | 0 | |||||||||||
| Virulence and Epitope prediction | ||||||||||||||||
| Protein ID | VirulentPred | VaxiJen Score | ElliPro Epitopes | |||||||||||||
| Linear | Discontinuous | |||||||||||||||
| D4YW28 | 1.0513 | 1.0137 | 5 | 12 | ||||||||||||
| Q8F3N1 | 1.0211 | 0.7507 | 3 | 8 | ||||||||||||
| Q8F1Y3 | 1.1107 | 0.7676 | 2 | 8 | ||||||||||||
| Q8F0U4 | 1.0804 | 0.7421 | 5 | 8 | ||||||||||||
| Q8EZJ1 | 1.0872 | 0.8526 | 3 | 12 | ||||||||||||
| Q8EY10 | 1.0471 | 0.7324 | 13 | 8 | ||||||||||||
| Protparam | ||||||||||||||||
| Protein ID | Size | Mol. Wt | PI | Negative residues | Positive residues | No. of atoms | Extinction coefficient | Instability index | Aliphatic Index | GRAVY | ||||||
| D4YW28 | 248 | 27451.22 | 9.91 | 26 | 45 | 3914 | 11460 | 44.08, unstable | 73.06 | -0.809 | ||||||
| Q8F3N1 | 178 | 19397.31 | 9.4 | 16 | 21 | 2751 | 10430 | 35.52, stable | 89.89 | -0.138 | ||||||
| Q8F1Y3 | 107 | 11797.32 | 6.31 | 18 | 18 | 1678 | 2980 | 37.93, stable | 78.41 | -0.816 | ||||||
| Q8F0U4 | 259 | 30150.65 | 8.84 | 35 | 39 | 4180 | 30830 | 47.95, unstable | 64.83 | -0.861 | ||||||
| Q8EZJ1 | 247 | 27324.03 | 9.57 | 31 | 42 | 3903 | 5960 | 54.47 Unstable | 80.93 | -0.603 | ||||||
| Q8EY10 | 439 | 49905.97 | 8.74 | 68 | 76 | 6926 | 89730 | 23.03 Stable | 48.18 | -1.134 | ||||||
| InterProScan | ||||||||||||||||
| Protein ID | Interpro Family ID | Description | ||||||||||||||
| D4YW28 | IPR030951 | Sec region non-globular protein | ||||||||||||||
| Q8F3N1 | IPR012902 | Prokaryotic N terminal methylation site | ||||||||||||||
| Q8F1Y3 | IPR031316 | Anti-sigma-28 factor FlgM superfamily | ||||||||||||||
| Q8F0U4 | None predicted | – | ||||||||||||||
| Q8EZJ1 | IPR036680 | Sporulation like domain superfamily | ||||||||||||||
| Q8EY10 | G3DSA:2.20.110.10 | Histone H3 K4 Specific Methyltransferase | ||||||||||||||
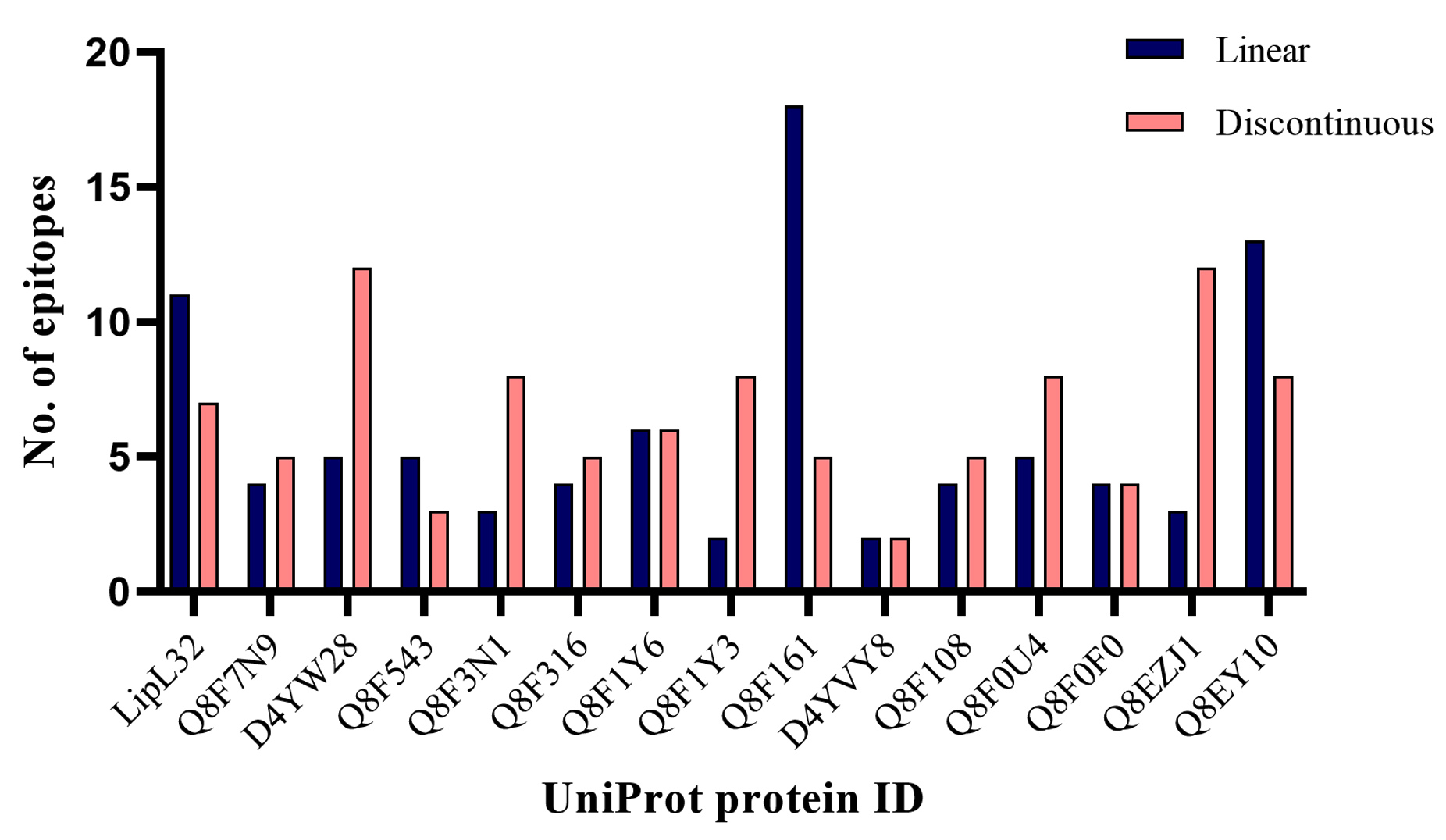
Figure 6. Graph representing the number of epitopes per classification in 14 significant proteins
Grouped column chart representation of Linear and discontinuous epitopes for 14 marker proteins and LipL32, predicted through ElliPro
A final list of proteins that qualified the epitope analysis arrived at six. The selected 6 proteins were screened through BepiPred 3.0. The epitope probability graph for all six proteins and LipL32 was given in Figure 7. The epitope regions were found to be proportioned as follows for each of the selected proteins with UniProt ID’s, D4YW28 (78%), Q8F3N1 (49%), Q8F1Y3 (64%), Q8F0U4 (62%), Q8EZJ1 (63%), and Q8EY10 (31%), and the data was represented as a pie chart in Figure 8. The proportion of epitope region in LipL32 was calculated to be 61%. Despite having 12 discontinuous epitopes, the epitope profile of Q8EZJ1 was found to be equivalent to LipL32, which left us with a single protein, D4YW28 that had a comparatively better epitope profile than LipL32.
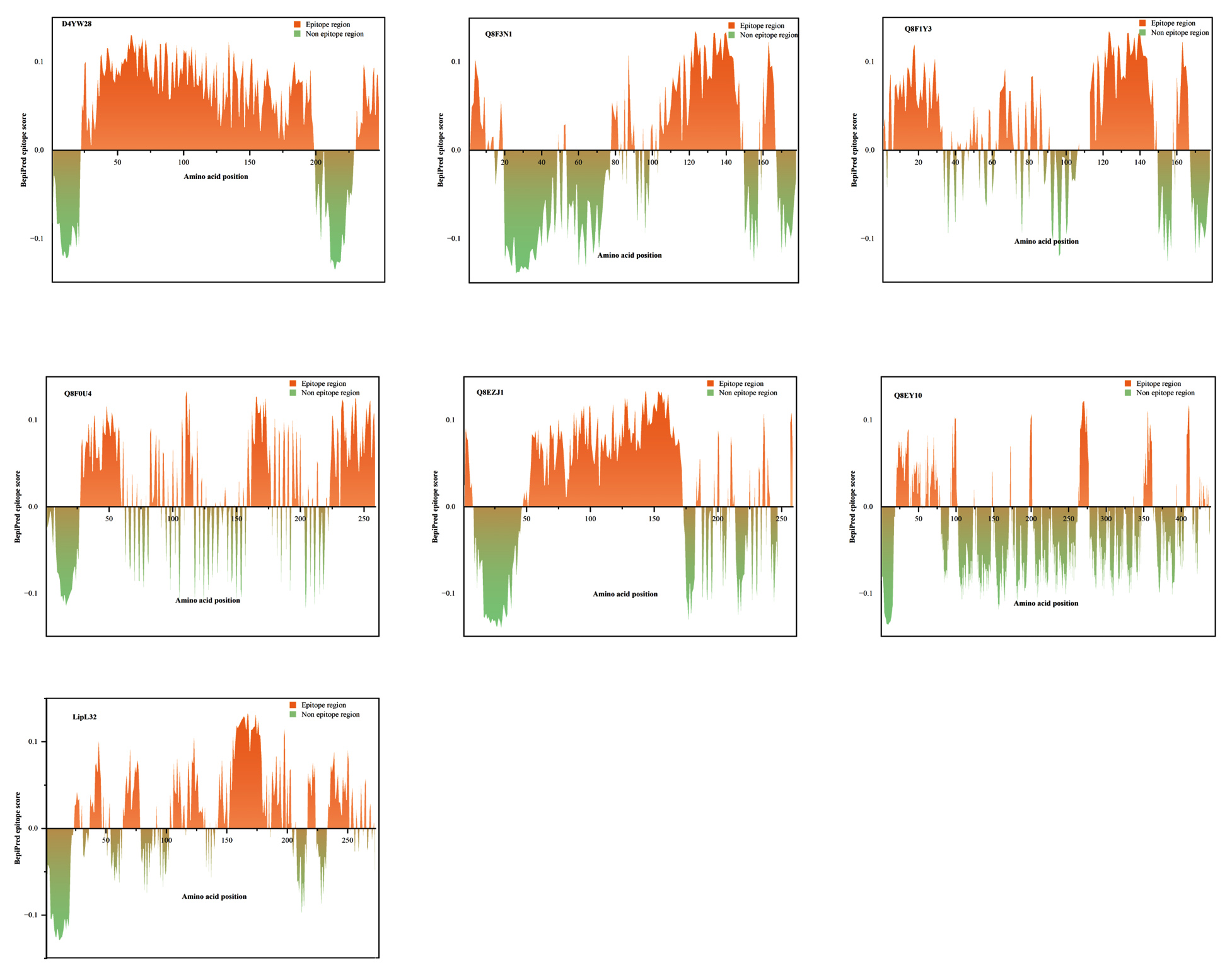
Figure 7. Epitope profiles of the potential biomarkers from BepiPred 3.0
Area chart representations of 6 significant proteins with better epitope profile compared to the standard biomarker LipL32. The x-axis represents the amino acid position and the y-axis represents the epitope score of the corresponding amino acid
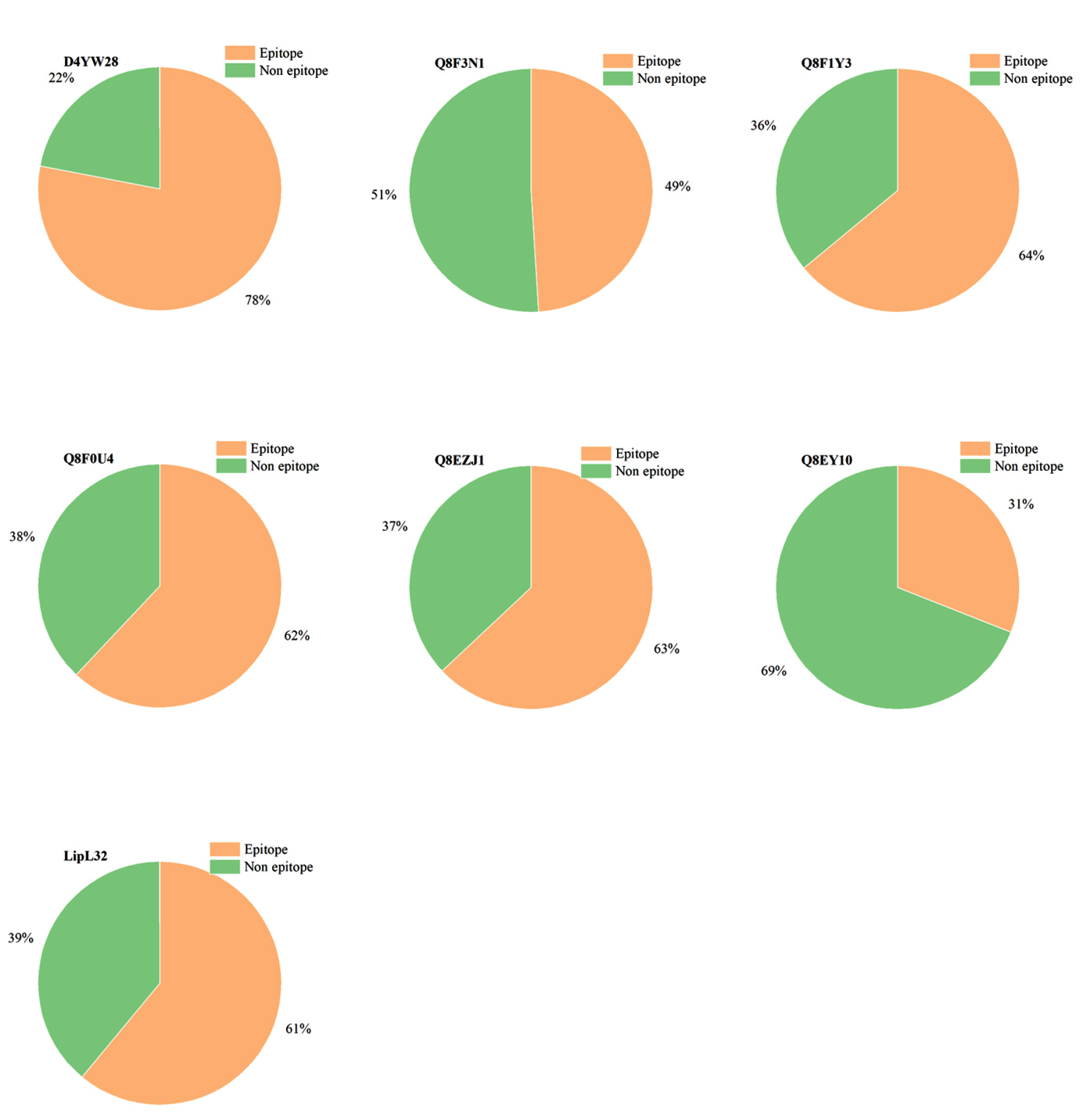
Figure 8. Representation of epitope and non-epitope regions as a pie chart in the significant marker proteins
Proportions of epitope and non-epitope regions in selected potential biomarkers are depicted in terms of a pie chart and compared with the standard marker, LipL32
The amino acids present in the regions 23-198 and 231-248 in D4YW28 gave higher epitope scores than the threshold fixed (0.1512) in BepiPred 3.0 (Figure 7). The protein, D4YW28 is of size 248 amino acids and out of those 194 amino acids possesses higher epitope scores contributing to the 78% epitope proportion which was greater than LipL32. The antigenic score determined by Vaxigen was way less for LipL32 (0.4789) compared to D4YW28 (1.0137). In discontinuous epitope prediction, Ellipro identified 7 epitopes for LipL32 and 12 for D4YW28. The discontinuous epitope regions of both the proteins are given in the Table S4 of Supplementary Material 2. The epitope profile of D4YW28 was better than other proteins which is possible because the discontinuous epitopes are distributed throughout the protein, the higher the number of discontinuous epitopes, the higher the chance for each amino acid to bind a paratope. The above analysis gestured to hypothesize that D4YW28 could serve as a better marker compared to LipL32. Noting its abundance in the leptospiral membrane, LipL32 was chosen to be a marker protein. Hence, the biomarker is expected to have better antigenicity but also be present abundantly in the organism. Using an overlapped peptide library, epitope mapping was done for LipL32, and the evaluation of epitopes was carried out by ELISA tests.41 Pre-predicted epitope regions of LipL32 were found to be in the location, 151-177 (Peptide-1), and 181-204 (Peptide-2). These experimental shreds of evidence were correlated with the epitope profile of LipL32 obtained from BepiPred 3.0 (present study), and the epitope scores for the amino acids present in the region 151-177 and 181-204, were higher than the threshold. Hence, it supports the hypothesis that the BepiPred data of D4YW28 is reliable too, making it eligible to be a diagnostic marker equivalent to LipL32. Conserved domains present in D4YW28 were checked in InterProScan where the protein is described under Sec-Region non-globular family found only in the genus Leptospira and the prediction is vouched by NCBI conserved domain database reflecting with an accession number, cl22800. The protein, D4YW28 is believed to be encoded between genes such as YajC and SecD Y. D4YW28 (https://www.uniprot.org/uniprotkb/D4YW28/) is found to possess a low-complexity sequence including Lys-rich and Ser/Thr/Asn/Glu-rich regions, stated in InterProScan.
The predicted biomarker needs thorough analysis before its exploitation in the diagnostic field. The establishment of D4YW28 as a biomarker needs strong evidence, such as information on its abundance and evaluation of its predicted epitope regions by ELISA or any other immunological assays. The abundance of the protein can be evaluated by conducting RT-PCR to quantify the mRNAs coding for D4YW28. Further, peptide mass fingerprinting (PMF) can be done by analyzing the peptide extracts from the L. interrogans through MALDI or ESI-MS (Electrospray Ionisation Mass spectrometry).42
Diagnosis of leptospirosis in the early stage has a great impact on the lifespan of the infected patients. The study reveals potential biomarkers for leptospirosis in the hypothetical protein population of L. interrogans serovar Lai. We unveiled the tendency of other proteins to serve as biomarkers that may overtake the standard leptospirosis biomarkers. The study found an extracellular protein D4YW28, having a greater epitope proportion and antigenic property among other candidates. The prediction tools were validated by analyzing LipL32, a standard biomarker for leptospirosis. This study suggests a direction for additional in vitro and in vivo research to assess the potential of the markers in vaccine development. Overall, our study provides the antigenicity profiles of significant proteins in L. interrogans which seek special attention to rule the diagnostic and vaccine field of leptospirosis.
Additional file: Additional Table S1-S3.
ACKNOWLEDGMENTS
The authors extend their special thanks to the Department of Genetic Engineering, SRM Institute of Science and Technology, Kattankulathur, Tamil Nadu, India, for providing research facilities and academic assistance.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
AUTHORS’ CONTRIBUTION
Both authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
FUNDING
None.
DATA AVAILABILITY
All datasets generated or analyzed during this study are included in the manuscript and/or in the supplementary files.
ETHICS STATEMENT
This article does not contain any studies on human participants or animals performed by any of the authors.
- Picardeau M, Bertherat E, Jancloes M, Skouloudis AN, Durski K, Hartskeerl RA. Rapid tests for diagnosis of leptospirosis: Current tools and emerging technologies. Diagn Microbiol Infect Dis. 2014;78(1):1-8.
Crossref - Rajapakse S. Leptospirosis: Clinical aspects. Clinical Medicine. 2022;22(1):14-17.
Crossref - Matthias MA, Lubar AA, Lanka ASS, et al. Culture-Independent Detection and Identification of Leptospira Serovars . Microbiol Spectr. 2022;10(6):e0247522.
Crossref - Haake DA, Levett PN. Leptospirosis in humans. Curr Top Microbiol Immunol. 2015;387:65-97.
Crossref - Levett PN. Leptospirosis. Clin Microbiol Rev. 2001;14(2):296-326.
Crossref - Bharti AR, Jarlath EN, Jessica NR, et al. Leptospirosis: A Zoonotic Disease of Global Importance. Lancet Infect Dis. 2003;3(12):757-771.
Crossref - Satiya J, Gupta NM, Parikh MP. Weil’s Disease: A Rare Cause of Jaundice. Cureus. 2020;12(6):e8428.
Crossref - Bhatt M, Rastogi N, Soneja M, Biswas A. Uncommon manifestation of leptospirosis: A diagnostic challenge. BMJ Case Rep. 2018;2018:bcr2018225281.
Crossref - Johnson RC. Leptospira. In Baron S, (eds.), Medical Microbiology, 4th Ed. University of Texas Medical Branch at Galveston (TX). 1996 (35) https://www.ncbi.nlm.nih.gov/books/NBK8451/
- Koizumi N, Picardeau M (eds.) Leptospira spp: Methods and Protocols. 1st Ed. 2020: 2134.
Crossref - Verma V, Kala D, Gupta S, et al. Leptospira interrogans outer membrane protein-based nanohybrid sensor for the diagnosis of leptospirosis. Sensors. 2021;21(7):2552.
Crossref - Matsunaga J, Barocchi MA, Croda J, et al. Pathogenic Leptospira species express surface-exposed proteins belonging to the bacterial immunoglobulin superfamily. Mol Microbiol. 2003;49(4):929-945.
Crossref - Picardeau M, Bulach DM, Bouchier C, et al. Genome sequence of the saprophyte Leptospira biflexa provides insights into the evolution of Leptospira and the pathogenesis of leptospirosis. PLoS One. 2008;3(2):e1607.
Crossref - Murray GL, Srikram A, Hoke DE, et al. Major surface protein LipL32 is not required for either acute or chronic infection with Leptospira interrogans. Infect Immun. 2009;77(3):952-958.
Crossref - Eisenstein E, Gilliland GL, Herzberg O, et al. Biological function made crystal clear-annotation of hypothetical proteins via structural genomics. Curr Opin Biotechnol. 2000;11(1):25-30.
Crossref - Pranavathiyani G, Prava J, Rajeev AC, Pan A. Novel Target Exploration from Hypothetical Proteins of Klebsiella pneumoniae MGH 78578 Reveals a Protein Involved in Host-Pathogen Interaction. Front Cell Infect Microbiol. 2020;10:109.
Crossref - Viratyosin W, Ingsriswang S, Pacharawongsakda E, Palittapongarnpim P. Genome-wide subcellular localization of putative outer membrane and extracellular proteins in Leptospira interrogans serovar Lai genome using bioinformatics approaches. BMC Genom. 2008;9:181.
Crossref - Marshall JS, Warrington R, Watson W, Kim HL. An introduction to immunology and immunopathology. Allergy Asthma Clin Immunol. 2018;14(Suppl 2):49.
Crossref - Nilvebrant J, Rockberg J. An introduction to epitope mapping. Methods Mol Biol. 2018;1785:1-10.
Crossref - Ferdous S, Kelm S, Baker TS, Shi J, Martin ACR. B-cell epitopes: Discontinuity and conformational analysis. Mol Immunol. 2019;114:643-650.
Crossref - Yu NY, Wagner JR, Laird MR, et al. PSORTb 3.0: Improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics. 2010;26(13):1608-1615.
Crossref - Yu CS, Chen YC, Lu CH, Hwang JK. Prediction of protein subcellular localization. Proteins: Struct Funct Genet. 2006;64(3):643-651.
Crossref - Imai K, Asakawa N, Tsuji T, et al. SOSUI-GramN: high performance prediction for sub-cellular localization of proteins in Gram-negative bacteria. Bioinformation. 2008;2(9):417-421.
Crossref - Kamper J, Kahmann R, Bolker M, et al. Insights from the genome of the biotrophic fungal plant pathogen Ustilago maydis. Nature. 2006;444(7115):97-101.
Crossref - Krogh A, Larsson B, Von HG, Sonnhammer ELL. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J Mol Biol. 2001;305(3):567-580.
Crossref - Wilkins MR, Gasteiger E, Bairoch A, et al. Protein identification and analysis tools in the ExPASy server. Methods Mol Biol. 1999;112:531-52.
Crossref - von Mering C, Jensen LJ, Snel B, et al. STRING: Known and predicted protein-protein associations, integrated and transferred across organisms. Nucleic Acids Res. 2005;33(Database Issue):D433-D4337.
Crossref - Tatusov RL, Fedorova ND, Jackson JD, et al. The COG Database: An Updated Version Includes Eukaryotes. BMC Bioinformatics. 2003;4:41.
Crossref - Salwinski L, Miller CS, Smith AJ, Pettit FK, Bowie JU, Eisenberg D. The Database of Interacting Proteins: 2004 update. Nucleic Acids Res. 2004;32(Database Isue):D449-451.
Crossref - Szklarczyk D, Gable AL, Lyon D, et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47(D1):D607-D613.
Crossref - Garg A, Gupta D. VirulentPred: A SVM based prediction method for virulent proteins in bacterial pathogens. BMC Bioinformatics. 2008;9:62.
Crossref - Doytchinova IA, Flower DR. Bioinformatic Approach for Identifying Parasite and Fungal Candidate Subunit Vaccines. The Open Vaccine Journal. 2008;1:22-26.
Crossref - Ponomarenko J, Bui HH, Li W, et al. ElliPro: A new structure-based tool for the prediction of antibody epitopes. BMC Bioinformatics. 2008;9:514.
Crossref - Clifford JN, Hoie MH, Deleuran S, Peters B, Nielsen M, Marcatili P. BepiPred-3.0: Improved B-cell epitope prediction using protein language models. Protein Sci. 2022;31(12):e4497
Crossref - Yang HL, Zhu YZ, Qin JH, et al. In silico and microarray-based genomic approaches to identifying potential vaccine candidates against Leptospira interrogans. BMC Genomics. 2006;7:293.
Crossref - Gamberini M, Gomez RM, Atzingen MV, et al. Whole-genome analysis of Leptospira interrogans to identify potential vaccine candidates against leptospirosis. FEMS Microbiol Lett. 2005;244(2):305-313.
Crossref - Yu CS, Lin CJ, Hwang JK. Predicting subcellular localization of proteins for Gram-negative bacteria by support vector machines based on n peptide compositions. Protein Sci. 2004;13(5):1402-1406.
Crossref - Magnus M, Pawlowski M, Bujnicki JM. MetaLocGramN: A meta-predictor of protein subcellular localization for Gram-negative bacteria. Biochim Biophys Acta. 2012;1824(12):1425-1433.
Crossref - Gillani M, Pollastri G. Protein subcellular localization prediction tools. Comput Struct Biotechnol J. 2024;23:1796-807.
Crossref - Raja V, Natarajaseenivasan K. Pathogenic, diagnostic and vaccine potential of leptospiral outer membrane proteins (OMPs). Crit Rev Microbiol. 2015;41(1):1-17.
Crossref - Lottersberger J, Guerrero SA, Tonarelli GG, Frank R, Tarabla H, Vanasco NB. Epitope mapping of pathogenic Leptospira LipL32. Lett Appl Microbiol. 2009;49(5):641-645.
Crossref - Paratsaphan S, Moonsom S, Reamtong O, et al. Characterization of a novel peptide from pathogenic Leptospira and its cytotoxic effect. Pathogens. 2020;9(11):906.
Crossref
© The Author(s) 2024. Open Access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License which permits unrestricted use, sharing, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.