


ISSN: 0973-7510
E-ISSN: 2581-690X
Serrapeptase is a proteolytic enzyme with many favourable biological properties like anti-inflammatory, analgesic, anti-bacterial, fibrinolytic properties and hence, is widely used in clinical practice for the treatment of many diseases. The activity of microbial enzymes is usually low and hence enhancing the enzyme activity is an integral and crucial area of research. In this study, a mutant with higher serrapeptase activity was developed by multiple exposures of Serratia marcescens to Ultra Violet radiation (UV) and Ethyl methyl sulfonate (EMS) individually and also a combination of both the methods was used. The mutant exposed only to UV radiation for 40 seconds indicated an increase in enzyme activity of 3234.9 EU/mL in comparison to wild-type strain (2770 EU/mL). The mutant exposed to only EMS indicated a very small increase in enzyme activity of 2797 EU/mL in comparison to wild-type strain (2770 EU/mL) but a lower enzyme activity than the UV mutant. Combinational exposure of wild-type to UV and EMS gave a better mutant with an enzyme activity of 3437.6 EU/mL which was higher than all the above methods. Optimization of temperature, incubation period and pH was studied in the wild type and mutant strains and found slight increase in activity in the mutant strain than the wild strain. The mutant serrapeptase has a molecular weight of approximately 50kDa and also exhibited fibrinolysis activity with a maximum blood clot lysis of 35% with 100 U/mL of Serrapeptase.
Serrapeptase, mutagenesis, EMS, UV radiation, fibrinolytic
Enzymes are biocatalysts and act as therapeutic agents for many disorders related to metabolism1. Microorganisms serve as an alternate source of enzymes in contrast to plants and animals2. Proteolytic enzymes are the class of enzymes that hydrolyze peptide bonds in proteins3 and hence play an important role in degrading protein aggregates in the body4. Serrapeptase (EC number 3.4.24.40) is one of the proteolytic enzymes belonging to serine protease family that were used by the ancient Japanese biochemist about 25 years ago and is widely used in medicine in the Asian and European countries5. It was initially isolated from the Enterobacteria Serratia marcescens strain E-15 that was found in the gut of the Japanese silkworm Bombyx mori. It has ability to dissolve only the dead proteins/non-living tissues6 and hence does not affect healthy tissues as the chemical structure of serrapeptase inhibits binding to proteins in healthy tissues7. It exhibits properties like antioedemic, analgesic, anti-inflammatory, dissolution of mucous, plaques and blood clots. Serrapeptase is effective in treating various diseases like atherosclerosis, arthritis, bronchitis, fibrocystic breast disease, crohn’s disease, breast engorgement, Alzheimer disease, hepatitis, lung disorders, diabetes, and uterine fibroids8.
Strain improvement of microorganisms by gene manipulations and genetic engineering can be easily performed using random mutation approaches or by recombinant DNA technology to produce mutant genes, proteins, bacterial strains or other genetically modified organisms and also to enhance enzyme activity and the rate of enzyme production9. By gene duplication and deletion mechanisms, mutations have to potential to enhance the efficiency of some genes. There is a variety of mutagenesis methods, few of them are physical methods like UV radiation, chemical mutagenesis mediated by chemicals like nitrous acid, EMS (Ethyl Methane Sulfonate), ethidium bromide etc. Physical mutagenesis methods like UV radiation induces mutations like frameshift, base substitutions, deletions, duplications and recombination10. When DNA is exposed to UV light (254nm/ 15W), DNA damage occurs resulting in pyrimidine dimers11. Ethidium Bromide inhibits nucleic acid biosynthesis by binding to it in-vitro and is an intercalating agent12. Methylnitronitrosoguanidine (MNNG) adds alkyl groups to the O6 of guanine and O4 of thymine creating a transition mutation between GC and AT. These changes cannot be detected by the DNA mismatch repair system as they do not create a heavy impression in the double helix of DNA. If the correct base is inserted, normal DNA sequence will be produced. Insertion of incorrect bases results in transversion or transition mutation. Another example is methyl nitrosoguanidine that adds methyl group to guanine causing it to miss pair with thymine. After subsequent replication, GC is converted into AT transition N-Methyl-N’-nitro-N-nitrosoguanidine (NTG) has been suggested to be the most potent chemical mutagen yet discovered13 and found that mutations to valine resistance and to auxotrophy occurred at high frequency than any others. EMS is the most commonly used chemical mutagen. It is an alkylating agent that causes point mutations, insertion or deletion mutations, mispairing of bases14 and causes mispairing like G pairs with T instead of C, resulting in G-C to A-T alterations15. EMS has the specificity to remove guanine and cytosine from the chain and results in gap formation. Any base (A, T, G, C) may be inserted in the gap.
In the present study, we attempted to improve the activity of wild-type serrapeptase enzyme by UV mutagenesis; EMS mediated mutagenesis and the combination of both the methods. First the wild-type S. marcescens strain MTCC8708 UV irradiated and treated with EMS separately and then combination of UV and optimal concentration of EMS was performed. Then the mutant strains were screened for protease activity on skim milk agar and activity for each mutant protein were found and compared. The effect of serrapeptase production on temperature, pH, and incubation period was studied and compared in the wildtype and final mutant strain. The serrapeptase produced from a selected mutant was concentrated, purified and characterized. The fibrinolytic potential of the mutant serrapeptase was also determined.
Chemicals
Serratia marcescens was purchased from MTCC, Chandigarh with the strain number 8708. EMS and BSA were purchased from SRL chemicals. Skim milk medium, LB broth, bacteriological agar, tryptone soya broth medium was purchased from Hi-Media. UV mutagenesis was carried out using UV cross-linker.
Culture conditions
S. marcescens live culture was plated on nutrient agar and incubated at 30°C for 24 hours. Single colony from the plate was inoculated into sterile nutrient broth and incubated at 37°C for 24 hours and stored at 4°C. It was maintained as 2 mL glycerol stocks in cryovials and kept at -80°C for long-term storage.
Random mutagenesis
For UV radiation mutagenesis of S. marcescens (OD 1.0), a UV crosslinker [UVP CX-2000 Crosslinkers] was used. The exposure was carried out at the distance 5 cm. The culture was plated in sterile nutrient agar medium plate and exposed to UV radiation of 15 W for different time intervals of 10 s, 20 s, 30 s, 40 s, 50 s, 60 s, 5 min, 10 min, 20 min, 30 min. The Petri plates were uncovered and occasionally mixed. After UV exposure these plates were wrapped in aluminium foil and were stored in dark.
Chemical mutagenesis was performed by taking 1ml of overnight culture with OD 1.0 and centrifuged at 10,000 rpm/min and the cells were washed and re-suspended in phosphate buffered saline (PBS).Different concentration of the EMS (5µg/mL to 250 µg/mL) was added to the bacterial suspension and incubated for 5min, 10 min, 30 min, 60 min, 90 min, 120 min and after each time interval the suspension is centrifuged and re-suspended in PBS16.
The mutant showing higher activity than the wild-type in both chemical and UV treatment is noted and the bacterial culture is exposed to UV radiation of 15 W for optimal time interval and then immediately treated with EMS of optimised concentration and time of exposure. The strain that is exhibiting the highest activity was selected for further studies.
Screening of wild and mutant strains for proteolytic activity
Casein hydrolysis method was employed here, in which skim milk agar medium was prepared and sterilized. A single line streak or a single dot of the S. marcescens pure isolate and mutants was made on skim milk agar and incubated at 37°C for 24 h. The plate was observed for a zone of clearance which confirms that the Isolate has proteolytic activity.
Serrapeptase production for wild and mutant strains
Single colony of S. marcescens wild and mutant strain was inoculated separately into 20 mL of sterile tryptone soya broth and incubated at 37°C and was monitored for an OD of 0.2-0.3 at 610 nm. 10 mL of the inoculum was transferred to 90 mL sterile production broth and incubated at 37°C for 24 hours in a shaker at 120 rpm/min. The medium was centrifuged at 10,000 rpm/min for 20 min at 4°C. The supernatant was filtered and stored at 4°C17.
Enzyme assay
The protease activity of SP was performed by using casein as substrate as mentioned in Sigmas Non-specific protease activity assay-casein as a substrate18. The protein content was determined by Bradford assay19 using BSA as standard.
Optimization of Serrapeptase production in wild and mutant strains
The effect of temperature on the activity of serrapeptase was determined by growing S. marcescens wild strain as well as the final mutant strain obtained through combined mutagenesis(UV+EMS)in the production medium and incubated it at different incubation temperatures such as 30°C, 35°C, 37°C and 40°C. The optimum incubation period was determined by growing S. marcescens wild strain as well as mutant strains in the production medium and incubated it at different incubation periods such as 6 h, 12 h, 18 h, 24 h, 30 h, 42 h and 48 h. The optimum pH was determined by adjusting the pH of the production medium using phosphate buffer prepared at different pH of 4,5,6,7,8,9, and 10. Serrapeptase was then extracted and the activity was analyzed by enzyme assay.
Serrapeptase purification
Ammonium sulfate fractionation
The cell free supernatant (crude) serrapeptase enzyme was first brought to 20% (w/v) saturation with solid ammonium sulfate according to the chart by Green and Hughes20. The precipitated proteins were separated by vortexing for 10 min followed by centrifugation for 10 min at 8000 rpm. The resultant pellet was dissolved in 5 ml of distilled water. The procedure was repeated for the left supernatant again with ammonium sulfate to achieve 40, 60, and 70% (w/v) saturation21. Both enzyme activity and protein content were determined for the precipitated fraction.
Ultrafiltration
The ammonium sulfate purified fraction was further subjected for ultrafiltration (Amicon Ultra-15, Millipore, India) with a 50 kDa molecular weight cut-off membrane and assayed for both protein content and Serrapeptase activity
Gel filtration chromatography
Gel filtration were carried out with a self-packed Sephadex G-100 column (1.6-cm in diameter; 25-cm gel bed height; 1-mL sample volume) equilibrated with sterile column buffer 0.01MTris–HCl and 0.01 M NaCl (pH7.5). Retentate (1 mL) collected from ultrafiltration was loaded on the prepared column with a flow rate of 0.5 mL/min. The fractions were eluted (3 mL), and protein content was analyzed at 280 nm. Further, the fractions containing protein content were checked for enzyme activity at 420 nm.
SDS PAGE
The SDS PAGE is done to determine the molecular weight of the serrapeptase and also to detect the presence of the desired enzyme in the lysate. The stacking gel was made at 5% and the separating gel was made at 12% acrylamide22. The staining solution contained methanol, water, glacial acetic acid in the ratio 5:4:1 with a pinch of coomassie brilliant blue dye in it. The destaining solution is same as staining solution with the exemption of the dye.
Fibrinolytic activity
The clot lysis assay was performed by taking 500μL of human blood in pre-weighed 1.5mL Eppendorf tubes and allowed it to sit undisturbed for an hour at 37°C for proper clotting to take place. The upper straw coloured layer is serum that is carefully aspirated out with a pipette without disturbing the clot. The tubes were again weighed to determine the weight of the clot formed. Then serrapeptase enzyme is added in different enzyme units (10EU/mL to 100 EU/mL) and incubated for 90minutes at 37°C to allow the clot lysis to happen23. After incubation, the dissolved blood is removed and again the tubes are weighed to determine the percentage of clot lysis that has occurred in each tube.
Statistical analysis
All experimentations were carried out in triplicates. Statistical significance was set at P values of ≤ 0.05. All graphs were prepared using the Graph Pad Prism-5 software.
Culturing of S. Marcescens wildtype
S. marcescens live culture obtained from MTCC was plated on nutrient agar and incubated at 30°C for 24 hours. Red colonies confirmed the culture obtained is S. marcescens (Fig. 1).
Fig. 1. Pure isolate of S. marcescens MTCC8708
Random mutagenesis
After UV mutagenesis of S. marcescens for different time intervals of 5s, 10s, 20s, 30s, 40s, 50s, 60s, 5min, 10min, 20min, 30min, 60min using 15W power, the bacteria could survive up to 30min and the maximum activity of the UV treated enzyme was observed for 40sec exposure with an enzyme activity of 3234.9 EU/mL (Fig. 2A) which is higher than the wildtype Serrapeptase (2770 EU/mL) and hence is considered as the best UV irradiated mutant.
S. marcescens culture was exposed to different concentration of EMS i.e. from 5µg/mL to 250 µg/mL for different time intervals i.e. 5min, 10 min, 30 min, 60 min, 90min and 120 min. It was found that the bacterial cells could resist the growth only up to 50 µg/mL for 10min. Hence 50µg/mL is the lethal concentration of EMS for S. marcescens. The maximum enzyme activity of 2797.6 EU/mL was observed for 30 µg/mL, 30 minutes incubation (Fig. 2B) and hence is considered as the best concentration-time combination for EMS mediated chemical mutagenesis.
After treating the bacterial culture with UV radiation for different time interval, they were subsequently treated with the optimized EMS concentration of 30 µg/mL, 30 minutes to further enhance the activity of the serrapeptase enzyme. The combination of 40 sec UV irradiation along with EMS treatment gave a maximum enzyme activity of 3437.6 EU/mL (Fig. 2C) which was greater than the wildtype enzyme as well as the best UV irradiated mutant. Hence this mutant was considered the best random mutant.
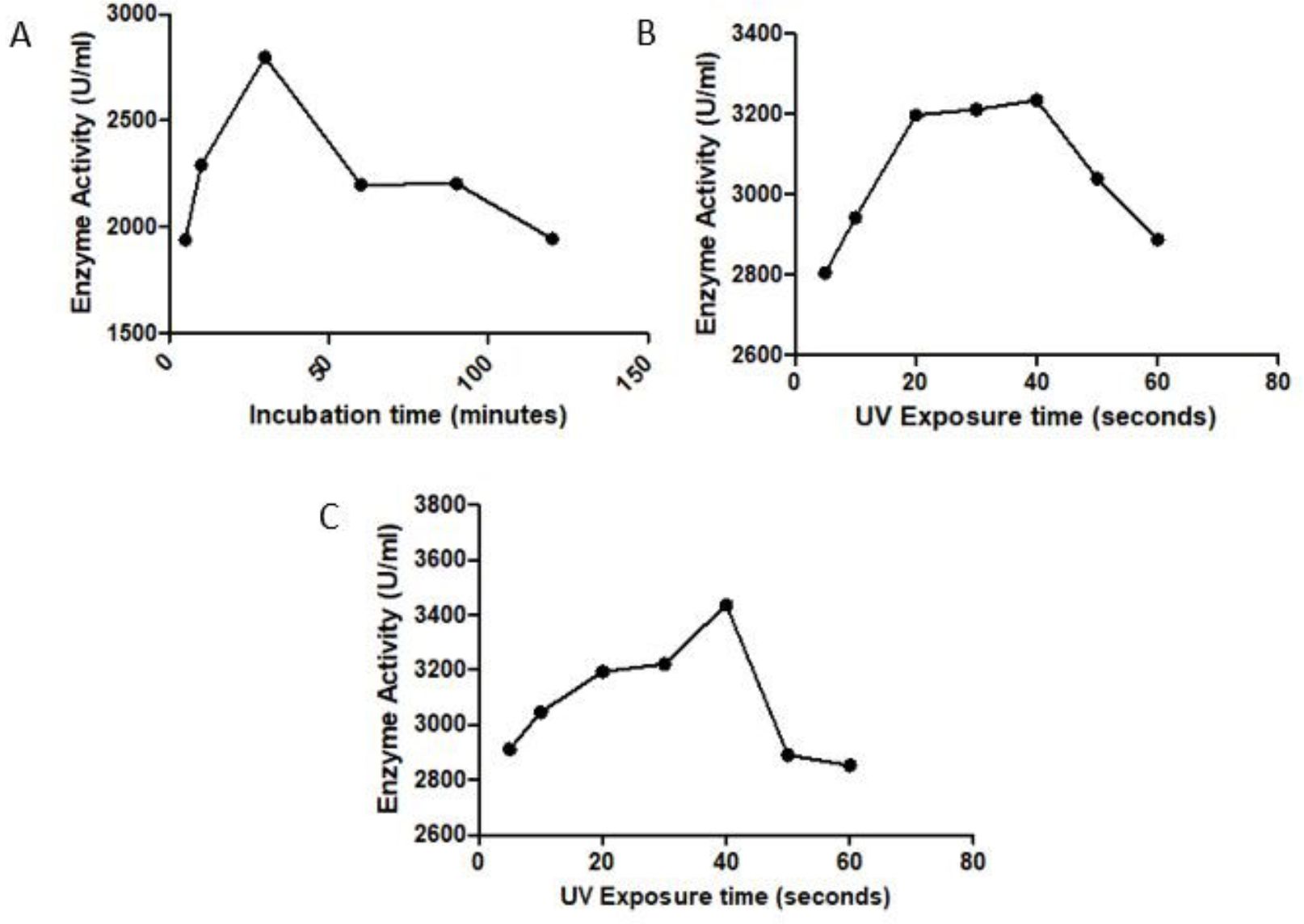
Fig. 2. Effect of random mutagenesis on serrapeptase activity: A) UV Mutagenesis, B) Chemical Mutagenesis, C) Combination of UV+EMS Mutagenesis
All the mutants were screened on skim milk for proteolytic activity and all the mutants showed clear zone indicating they possess proteolytic activity (Fig. 3)
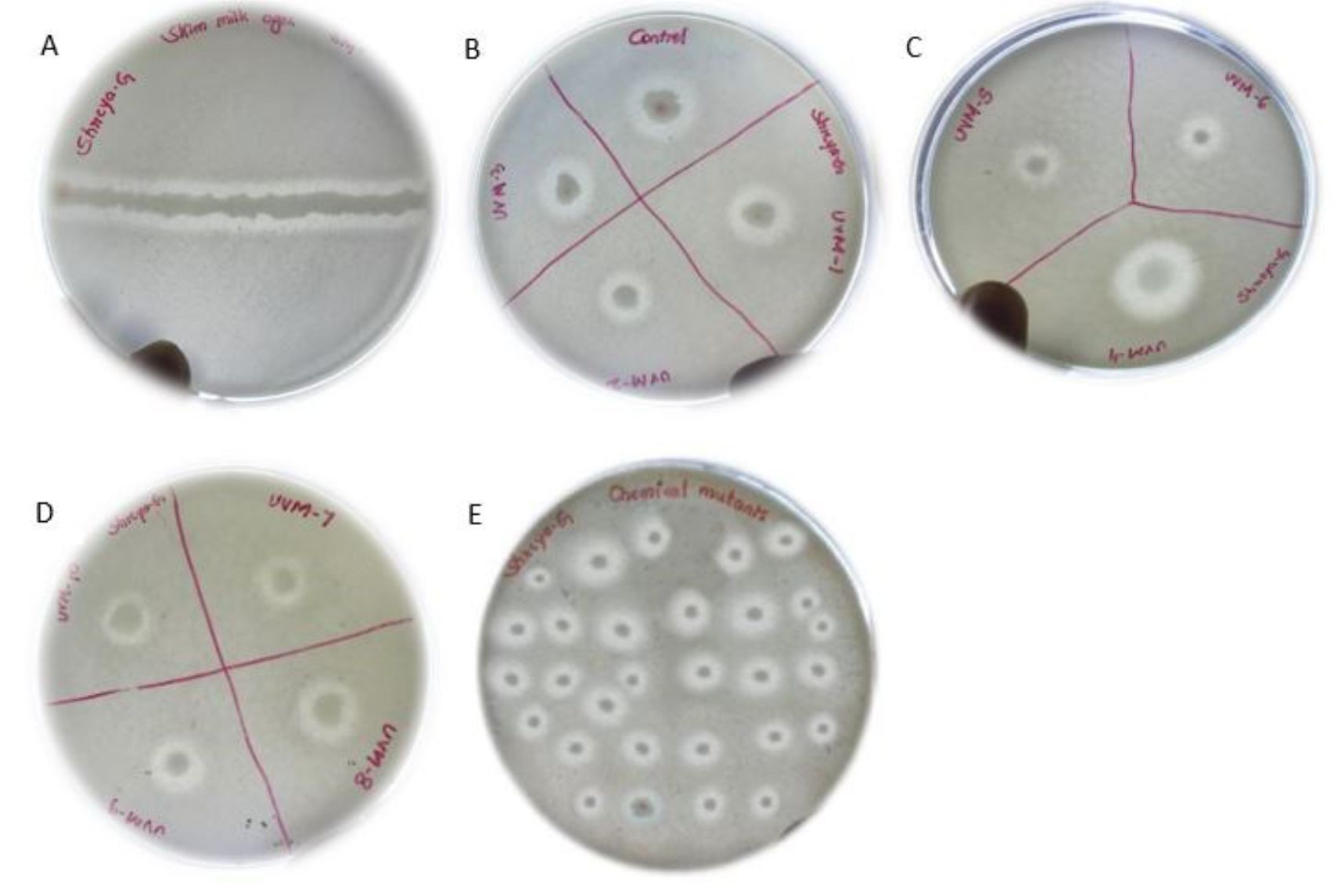
Fig. 3. Screening for proteolytic activity. (A) wild-type B) control (wild type) and S. marcescens UV mutants-1 (10s), UVM-2 (20s), UVM-3 (30s). ( C) UVM-4 (40s), UVM-5 (50s), UVM-6 (60-s), (D) UVM-7 (5min), UVM-8 (10min), UVM-9 (20min), UVM-10 (30min) and (E) EMS treated mutants, and UV+EMS treated mutants.
The S. marcescens wild strain and mutant strain was inoculated in production medium at different incubation temperatures such as 30°C, 35°C, 37°C and 40°C, the medium was centrifuged at 10,000 rpm and the supernatant (Serrapeptase enzyme) was collected and protease assay was performed. Tyrosine was used to obtain the standard curve. The maximum enzyme activity of 2770 EU/mL and 3582.10 EU/mL was observed at 37°C in the wild and mutant Serrapeptase respectively (Fig. 4A) and hence considered the optimum temperature for Serrapeptase production.
After incubating the production medium of wild and mutant strain for different incubation periods such as 6 h, 12 h, 18 h, 24 h, 30 h, 42 h, and 48 h, the medium was centrifuged at 10,000 rpm and the supernatant (Serrapeptase enzyme) was collected and protease assay was performed. The maximum enzyme activity of 2720 EU/mL and 3537.633EU/mL was observed at 24 hours in the wild and mutant Serrapeptase respectively (Fig. 4B) and hence considered the optimum incubation period for Serrapeptase production.
The optimum pH was determined by adjusting the pH of the production medium of wild and mutant strain to different pH of 4,5,6,7,8,9,10 and the maximum enzyme activity of 2171.0 EU/mL and 2812.14EU/mL was observed at pH 7 in the wild and mutant Serrapeptase respectively (Fig. 4C) and hence it is considered as optimum pH for Serrapeptase production.
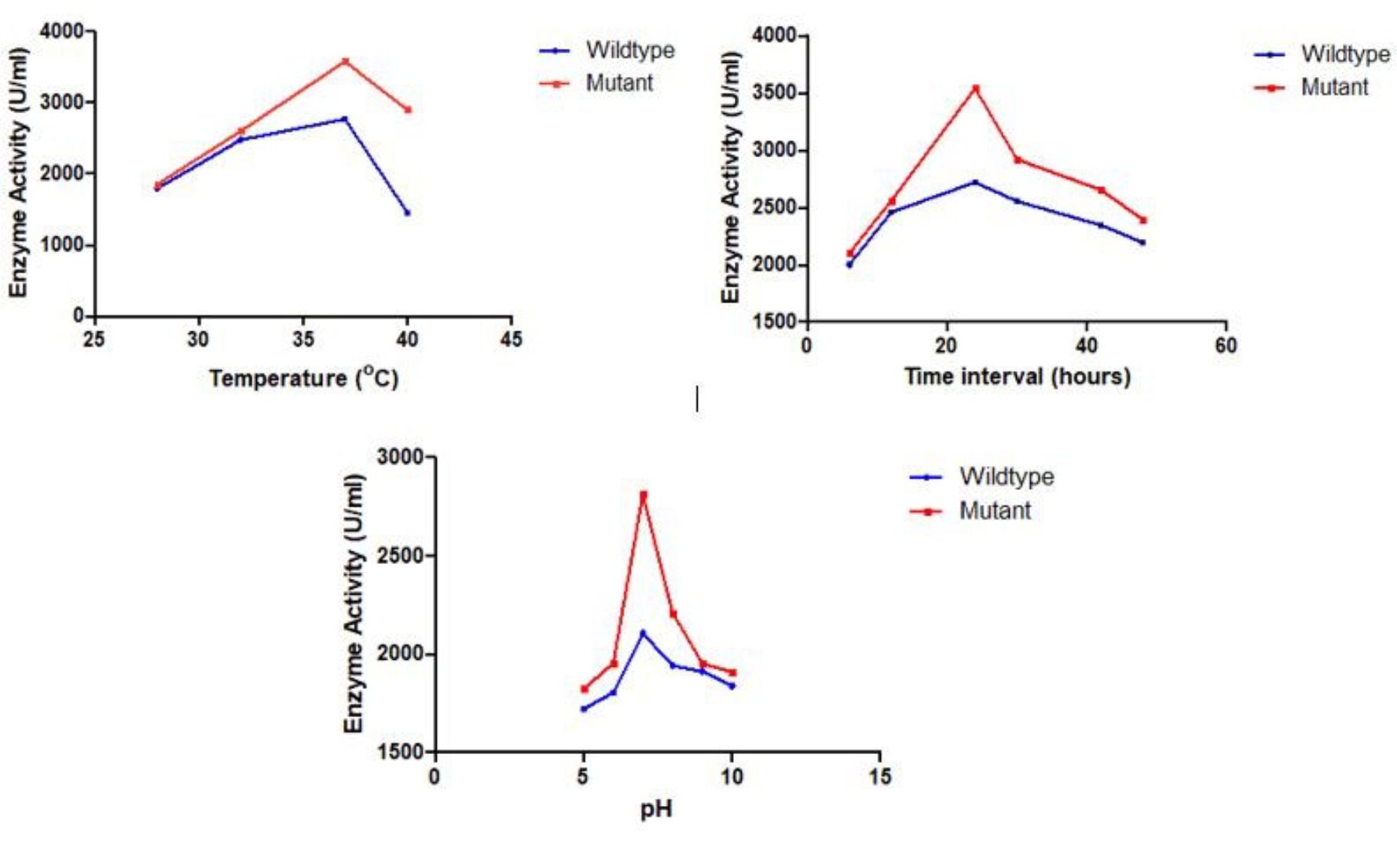
Fig. 4. Optimization of Serrapeptase production for wild and mutant strains on different parameters A) Temperature, B) Incubation period, and C) pH.
Hence the optimum conditions for mutant Serrapeptase production was found to be temperature of 37°C , pH of 7, and 24 hours of incubation period.
Serrapeptase purification
The UV+Chemical treated serrapeptase was selected as the best random mutant based on activity of Serrapeptase. This enzyme was partially purified by ammonium sulfate purification and the protein was precipitated at a saturation of 70% ammonium sulfate. This precipitate was dissolved in 5ml of distilled water. The ammonium sulfate purified fraction was further filtered through 50kDa amicon filter and the raffinate phase was further subjected to gel permeation chromatography using Sephadex and the fraction in which serrapeptase was present was determined using SDS PAGE. The protein content and enzyme activity was determined for all the purification steps and purification fold was calculated for samples (Table 1).
Table (1):
Serrapeptase purification.
Protein fraction |
Protein Content (mg/mL) |
Activity (units/mL) |
Specific activity (units/mL) |
Purification fold |
|---|---|---|---|---|
Crude |
10.7 |
3582.10 |
334.77 |
1 |
Ammonium sulfate |
11.3 |
7468.73 |
660.95 |
1.97 |
Ultrafiltration |
11.5 |
10794.82 |
938.68 |
2.80 |
Size exclusion chromatography |
12.3 |
17877.31 |
1453.44 |
4.34 |
Fibrinolytic assay
The clot lysis assay was performed by forming blood clot in 1.5mLEppendorf tubes and then treating the clots with serrapeptase enzyme from 10 EU/mL to 100 EU/mL. In Fig. 5, we can clearly see there was blood (liquid) that was getting collected below the tube indicating lysis has occurred due to the action of Serrapeptase. The weight of clot before and after lysis gives the % of clot lysis for each enzyme units as shown in the graph (Fig. 6). Clearly there was an increase in the lysis percentage as the enzyme units increased. The maximum clot lysis of 35% was observed when 100 units of enzyme was used.
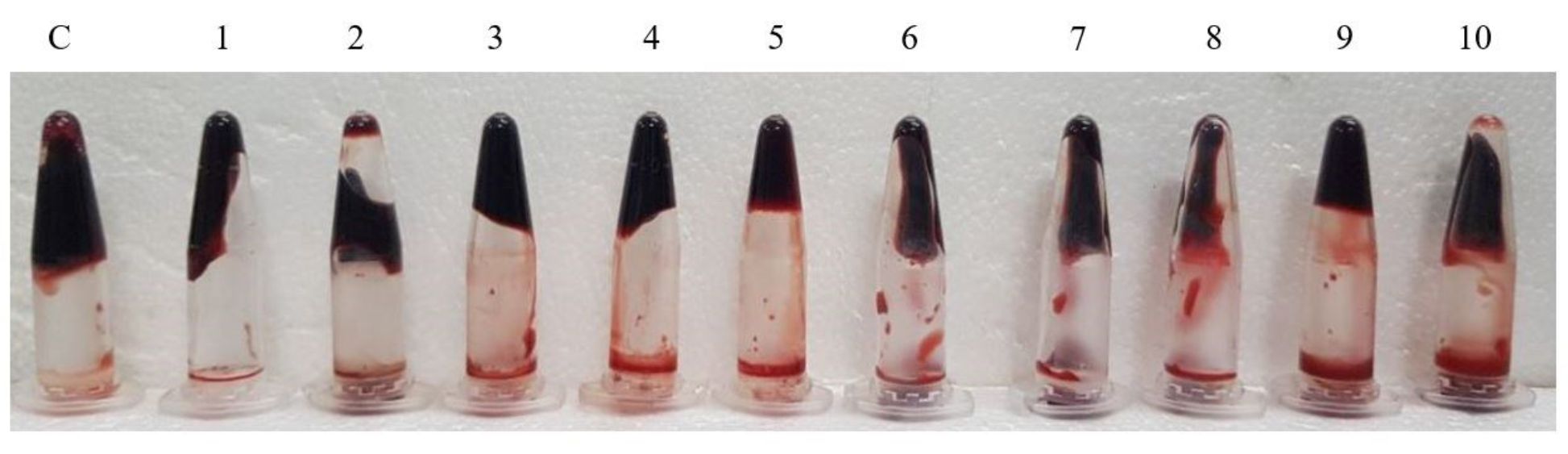
Fig. 5. Clot lysis assay for Serrapeptase. C: control (treated with water), 1-10: Serrapeptase enzyme treated from 10U/mLto 100 U/mL.
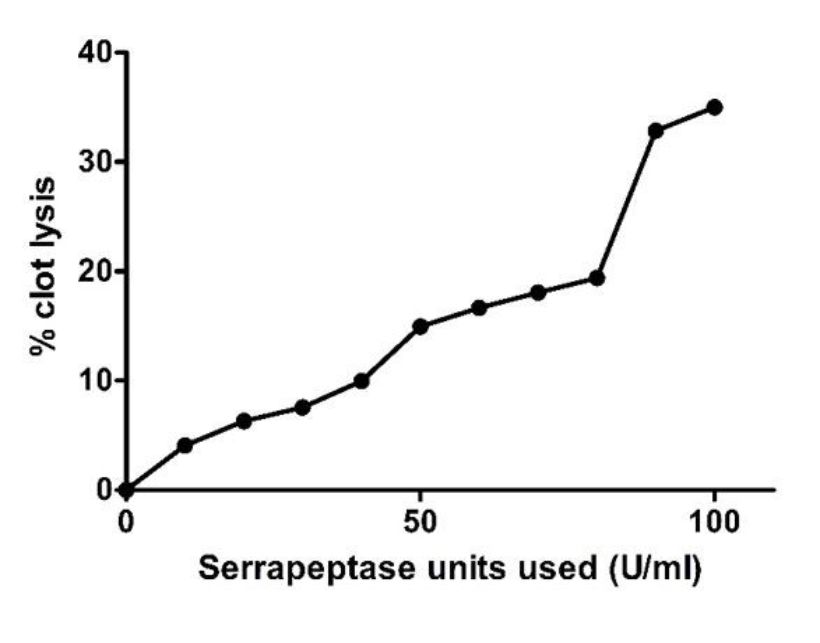
Fig. 6. Effect of Serrapeptase on blood clot lysis
In this study, we optimized the conditions for production of serrapeptase enzyme from S. marcescens. Random mutagenesis approach was selected for strain improvement to enhance the activity of the serrapeptase. UV irradiation was selected as physical mutagenesis method and Ethyl methane sulfonate (EMS) was used as a chemical mutagen for chemical mutagenesis. A combination of both UV and EMS was also done to further enhance the activity.
Various studies on optimization of serrapeptase have been done earlier with this enzyme. In this study, the optimum conditions for Serrapeptase production was found to be a temperature of 37°C, pH of 7, 24 hours of incubation period, 1% casein as substrate concentration, tryptone as best nitrogen source and sucrose as best carbon source which were quite similar to the earlier published results. A study by Manal , found the optimum temperature for serrapeptase production to be 32°C, incubation time of 24 hours with supplement of maltose as an carbon source24. They did not compare the results with different carbon sources in their study and hence their result is accurate for maltose as carbon source. While Mohankumar and Raj, found the optimum temperature to be 37°C, pH to be 7.317. Anil et al. found the optimum incubation period for serrapeptase production to be 25hours25 while Mohankumar and Raj, found it to be 24 hours17. The variation in each study may be due to the strain of Serratia used and also the laboratory conditions.
UV mutagenesis study indicated that exposure to UV radiation could increase or decrease the enzyme activity. An UV exposure of 40 seconds gave a maximum enzyme activity of 3234.9 EU/mL which is higher than the wildtype enzyme activity of 2770 EU/mL. EMS treatment resulted with a maximum enzyme activity of 2.797 EU/mL which is very slightly higher than the wild-type. But a combination of UV and EMS gave an enzyme activity of 3437.6 EU/mL which is the highest of all the approaches. Hence combination of UV and EMS lead to further enhancement of enzyme activity. The study conducted by Manal increased the production of serrapeptase with a UV exposure time of 20seconds while there was no effect observed with the further increase in UV exposure time.
Strain improvement was performed by random mutation using the UV and EMS, and also combination of both. The UV and EMS combinational method of strain improvement method produced a new mutant strain which has properties of producing more amount of Serrapepatse enzyme than the wildtype strain and also has higher activity when compared to the wildtype enzyme. Optimization of the carbon, nitrogen source, pH, incubation time, and temperature also increased the yield of the serrapeptase enzyme.
ACKNOWLEDGMENTS
The authors would like to thank SRM IST, Chennai, India for supporting and performing the study.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
AUTHORS’ CONTRIBUTION
ES and SG designed all the experiment. SG and RV drafted the manuscript, compiled information from the literature, and designed the Figs. and tables. ES supervised and reviewed the manuscript. KNR and SD did protein purification, English language editing and proof reading.
FUNDING
None.
ETHICS STATEMENT
Not applicable.
AVAILABILITY OF DATA
None.
- Cech TR, Bass BL. Biological catalysis by RNA. Annu Rev Biochem. 1986;55(1):599-629.
Crossref - Singh R, Kumar M, Mittal A, Mehta PK. Microbial enzymes: industrial progress in 21st century. 3 Biotech. 2016;6(2):174.
Crossref - Bach E, Anna SV, Daroit DJ, Correa APF, Segalin J, Brandelli A. Production, one-step purification, and characterization of a keratinolytic protease from Serratia marcescens P3. Process Biochem. 2012;47(12):2455-2462.
Crossref - Jickling GC, Zhan X, Ander BP, et al. Genome response to tissue plasminogen activator in experimental ischemic stroke. BMC Genomics 2010;11(1):254.
Crossref - Selvarajan E, Shreya Gopinath. Production, purification, characterization, immobilization, and application of Serrapeptase: a review. Frontiers of Biology in China. 2017;12(5):333-348.
Crossref - Sellman S. Serrapeptase, An Amazing Gift from The Silk Worm. World rights reserved, 2003.
- Robert SR. The ‘Miracle’ Enzyme is Serrapeptase, the 2nd Gift from Silkworms Giving the answer to Pain, Inflammation and Clear Arteries. Naturally Healthy Publications. 2009;3(4):12-19.
- Klein G, Kullich W. Short-term treatment of painful osteoarthritis of the knee with oral enzymes: randomised, double blind study versus Diclofenac. Clin Drug Invest. 2000;19(1):15-23.
Crossref - Illanes A, Cauerhff A, Wilson L, Castro GR. Recent trends in biocatalysis engineering. Bioresour Technol. 2012;115(1):48-55.
Crossref - Schaaper RM, Dunn RL, Glickman BW. Mechanisms of ultraviolet induced mutation: mutational spectra in Escherichia coli lacI gene for wildtype excision-repair-deficient strain. Journal of Molecular Biology. 1987;198(2):187-202.
Crossref - Wehner J, Horneck G. Effects of vacuum UV and UVC radiation on dry Escherichia coli plasmid pUC19: I. Inactivation of lacZ − mutation induction and strand breaks. Journal of Photochemistry and Photobiology. 1995;28:77-85.
Crossref - Bishop PE, Brown LR. Ethidium bromide resistant mutant of bacillus subtilis. J Bacteriol. 1973;115(3):1077-1083.
- Adelberg EA, Mandel M, Chen GCC. Optimal conditions for mutagenesis by N-methyl-N’-nitro-N-nitrosoguanidine in Escherichia coli K12. Biochem Biophys Res Commun. 1965;18:788-795.
Crossref - Gayou O, Aniol KA, Averett T, et al. Measurement of G E p/G M p in e→p→e p→to Q 2= 5.6 GeV 2. Phys Rev Lett. 2002;88(9):092301.
Crossref - Walker GC. Mutagenesis and inducible responses to deoxyribonucleic acid damage in Escherichia coli. Microbiol Rev. 1984;48:60-93.
Crossref - Bose JL. Chemical and UV mutagenesis. In The Genetic Manipulation of Staphylococci. 2014; 111-115. Humana Press, New York, NY.
Crossref - Mohankumar A, Raj RHK. Production and characterization of Serratiopeptidase enzyme from Serratia marcescens. Int J Biol. 2011;3(3):39-51.
Crossref - Cupp EC. Sigma’s Non-specific Protease Activity Assay – Casein as a Substrate. J Vis Exp. 2008;19:899.
Crossref - Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248-254.
Crossref - Green AA, Hughes WL. Protein solubility on the basis of solubility in aqueous solutions of salts and organic solvents. Methods Enzymol. 1955;1:67-90.
Crossref - Salarizadeh N, Hasannia S, Noghabi KA, Sajedi RH. Purification and Characterization of 50 kDa Extracellular Metalloprotease from Serratia sp. ZF03. Iran J Biotech. 2014;12(3):18-27.
Crossref - Machielsen R, Uria AR, Kengen SWM, Van OJ. Production and characterization of a thermostable alcohol dehydrogenase that belongs to the aldo-keto reductase superfamily. Appl Environ Microbiol. 2006;72(1):233-238.
Crossref - Sweta P,Rajpal SK, Jayant YD, Hemant JP, Girdhar MT, Hatim FD. Development of an in vitro model to study clot lysis activity of thrombolytic drugs. Thromb J. 2006;4:14.
Crossref - Manal KM. Effect of temperature and mutation on serratiopeptidase secreted from Serratia marcesence. Journal of Genetic and Environmental Resources Conservation. 2015;3(1):35-37.
- Anil CS, Kashinath MA. Production, Characterization and optimization of potent protease (Serratiopeptidase) from Serratia marcescens E 15. Int Res J Pharm App Sci. 2013;3(3):95-98.
© The Author(s) 2020. Open Access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License which permits unrestricted use, sharing, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.