


ISSN: 0973-7510
E-ISSN: 2581-690X
Vabeiryureilai Mathipi1![]() , Surajit De Mandal1
, Surajit De Mandal1![]() , Zothansanga Chawngthu2, Ruth Lalfelpuii1
, Zothansanga Chawngthu2, Ruth Lalfelpuii1![]() , Nachimuthu Senthil Kumar1
, Nachimuthu Senthil Kumar1![]() and H. Lalthanzara2
and H. Lalthanzara2 ![]()
Earthworms are important members of the soil macrofauna that play a significant role in soil structure and fertility. However, there is scanty information on the earthworm gut microbial flora and their metabolic potential. In the present study, the diversity of the microbial community and their metabolic potential from the gut content of four different earthworm species from Indo-Myanmar Biodiversity Hotspot were collected and identified by standard methods. The microbial diversity and their metabolic potential were assessed by high throughput sequencing of V3-V4 region of 16S rRNA using Illumina technology. Analysis of microbial diversity was performed by QIIME software package v.1.8.0 with their metabolic potential by PICRUSt (v1.1) software package. A total of 3,36,047 processed sequences were obtained that generated 3686 operational taxonomic units (OTUs). Major bacterial phyla identified were Proteobacteria (47.1%), Firmicutes (38.9%), Actinobacteria (6.3%), Bacteroidetes (3.6%) and Cyanobacteria (1.1%). The abundant genera were Lysinibacillus (26.9%), Acinetobacter (21.2%), Pseudomonas (4.7%), Bacillus (3.8%), Staphylococcus (3.5%), Stenotrophomonas (1.1%) and Ralstonia (1%). The functional annotation of the metagenome revealed abundance of bacterial community associated with amino acid, carbohydrate as well as energy metabolism. Furthermore, the presence of enzymes involved in the process of denitrification and methanogenesis were also identified. This study gives insight into the gut microbial composition and their putative functional roles in the gut of tropical hilly earthworms. The study on forest and garden soil earthworm gut microbiomes might help us understand the role of these organisms in their respective ecosystems.
Earthworm gastrointestinal, microbial, Mizoram, Next Generation Sequencing, soil
Earthworms are known as ‘keystone species’ in soil food webs and play an important function in soil nutrient cycling1. The major portion of the invertebrate biomass present in the soil is contributed by earthworm2. During earthworm’s feeding, the microorganisms were also ingested along with organic matter for their nutrients3. The microbiota present inside the earthworm gut is the main driver of their beneficial activities4-5. Most of the gut microbes harboured by earthworms are acquired from their surrounding habitats and perform different activities including fermentation and denitrification6-8. The capacity of the microorganisms to endure the enteric condition of the gut of earthworm is very important3. The ingestion of microorganism’s populations plays a major role in earthworm’s nutrition by facilitating in the decomposition of organic materials, mostly the constituents which are the earthworms cannot utilize in their natural condition9. The oxygen-limited environment prevalent in the earthworm gut allows the ingested anaerobes to grow and secrete exoenzymes which help the degradation process of the complex organic materials7,10.
The relationship between earthworms and microorganisms is at the level of their digestive tract, castings, and burrow walls2. They maintain mutualism that exists between earthworms and microorganisms11 along with higher microbial activity in earthworm castings. The plant nutrient availability in soil is likely relying on the activity of earthworm gut microflora and the diversity of the microorganisms depends on the biotic environment along with various features such as temperature, humidity, apparent density, pH, and organic matter which form part of their nourishment12. The member of the Rhizobiale bacteria was involved in the denitrification process and produces N2O gas inside earthworm gut5,7,13. Earthworms are known to have an association with free-living soil bacteria and constitute the drilosphere14-15. The gut of earthworms Lumbricus terrestris and Aporectodea caliginosa are reported to comprise a higher number of aerobes compared to the soil16.
Majority of the gut metagenomic studies focus on the epigeic species, whereas anecic earthworms gut microbial study is still scanty10. In the Indian subcontinent, only a handful of work has been carried out on microbes of earthworm gut content. A good number of papers regarding bacterial diversity of earthworm alimentary canal was published17-20. The report on the taxonomic with functional annotation of gut microbial communities of two epigeic earthworm species (Eisenia foetida and Perionyx excavatus)21 as well as an exploration of epigeic earthworm gut micro-biome in respect to detoxification of nanoparticles in soil system are also published22. Isolation and characterization of gut microflora of an epigeic P. excavatus was reported from West Bengal23. The common Indian earthworm Lampito mauritii was also assessed for its gut bacteria community24. The recent study on microbial diversity of an earthworm gut was done from the surface dwellers region only (epigeic species) like E. fetida, Eudrilus eugeniae and P. excavatus at the global level, and an epi-anecic species like L. terrestris and A.caliginosa of the temperate region only25,26. There is no work on gut biota of tropical anecic earthworms.
Considering the scarcity of information on the earthworm gut microbial communities in tropical hilly regions and in particular from Indo-Myanmar Biodiversity hotspot region, the present study aims to characterize the microbial diversity, community profile and their putative metabolic potentials of the gut microbiota of an anecic and an epi-endogeic species of earthworms.
Collection, identification and extraction of earthworm gut content
The earthworm sampling was done by digging and hand sorting method. The earthworm specimens were collected from brownish-grey loamy and light grey sandy garden soil with slightly acidic pH (5.8 – 6.4) and 22-25% moisture content in Aizawl city in Mizoram, northeast India (Indo-Myanmar biodiversity hotspot) and the worms were transported to the laboratory for identification.
The earthworm specimens were identified by Lalthanzara in consultation with monographs27-29. One individual each of four species of earthworms such as Eutyphoeus gigas (Stephenson), Eutyphoeus sp., Amynthas alexandri (Beddard) and Metaphire houlleti (Perrier) were selected (Table 1). For gut content analysis, the live earthworms were washed thoroughly, sacrificed and the skin was cut open to expose the gut from the dorsal site without puncturing the alimentary canal. The intestine was carefully cut open from post-clitellum to posterior end with a fine sterile blade following dissection method23,30-31. Maximum precaution was taken to avoid any contamination with tissues and other contaminants. The intestinal content was collected into a sterilized Eppendorf tube and refrigerated at -20˚C for further microbial analysis.
Isolation of metagenomic DNA and sequencing
The metagenomic DNA was isolated from gut samples by using the Fast DNA spin kit (MP Biomedical, USA) and measured quantitatively by a microplate reader (Spectra Max 2E, Molecular Devices, USA). We employed high throughput Illumina sequencing and software tools to survey the microbial community and its potential putative functions of the earthworm species along the elevation gradient of a mountainous place in the Indo-Myanmar biodiversity hotspot region. The V3-V4 hyper variable region of the 16S rRNA gene was sequenced using Illumina MiSeq (Illumina Inc., San Diego, CA, USA).
The QIIME software package (v.1.8.0) was used to process and analyze raw fastq sequences32-33. Sequences quality score <25 with reading length <200 bp were filtered and chimeric sequences were removed using USEARCH34-35. Preprocessed V3-V4 sequences were assembled into operational taxonomic units (OTUs) using the Uclust (similarity cutoff = 0.97)36. Each OTU of their representative sequence was classified using Green genes database37-38. PICRUSt (v1.1) (Phylogenetic Investigation of Communities by Reconstruction of Unobserved States) was used for the identification of phylogenetic differences among the microbial communities of earthworms on the metabolic potential following the recommended workflow. The functional composition of the metagenome was predicted by using marker gene data (16S rRNA) along with the genomes reference database39.
Characterization of microbial diversity
A total of 3,36,047 filtered sequences are obtained from the raw sequences of four earthworm species and a total of 3686 OTUs were obtained. The good’s coverage of the samples for 16SrRNA amplicon was found to be 99.71% (PUCZM375A), 99.84% (PUCZM351), 99.86% (NRY1) and 99.92 % (EWBK2) (Mean ± SD) which implied that most of the diversity were captured. The estimation of alpha diversity indices revealed a significant difference in bacterial communities of earthworm gut samples (between pH, habitat type and altitude) (Fig. 1). The species richness was highest in PUCZM375A (1060.686) followed by PUCZM351 (981.647), NRY1 (7584.264) and EWBK2 (527.882). A significant variation was also detected in the non-parametric Shannon index for microbial population and ranged from PUCZM351 (2.338) PUCZM375A (3.154), EWBK2 (4.882) and lowest in NRY1 (6.990) (Fig. 1). In this study, we found a significant effect of the type of earthworm species on the beta diversity of the microbiome, which was evident using UniFrac distances (Fig.2).
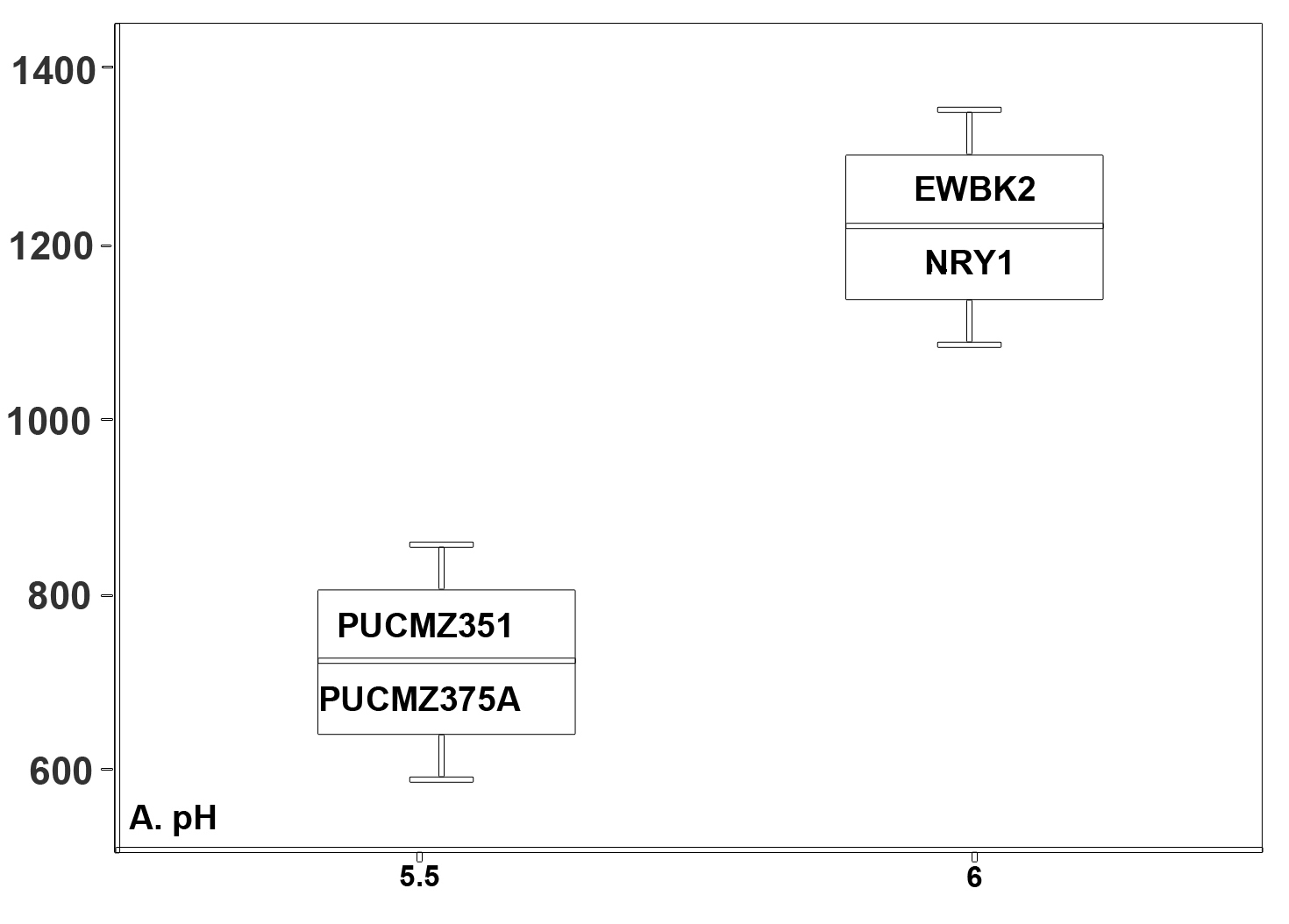

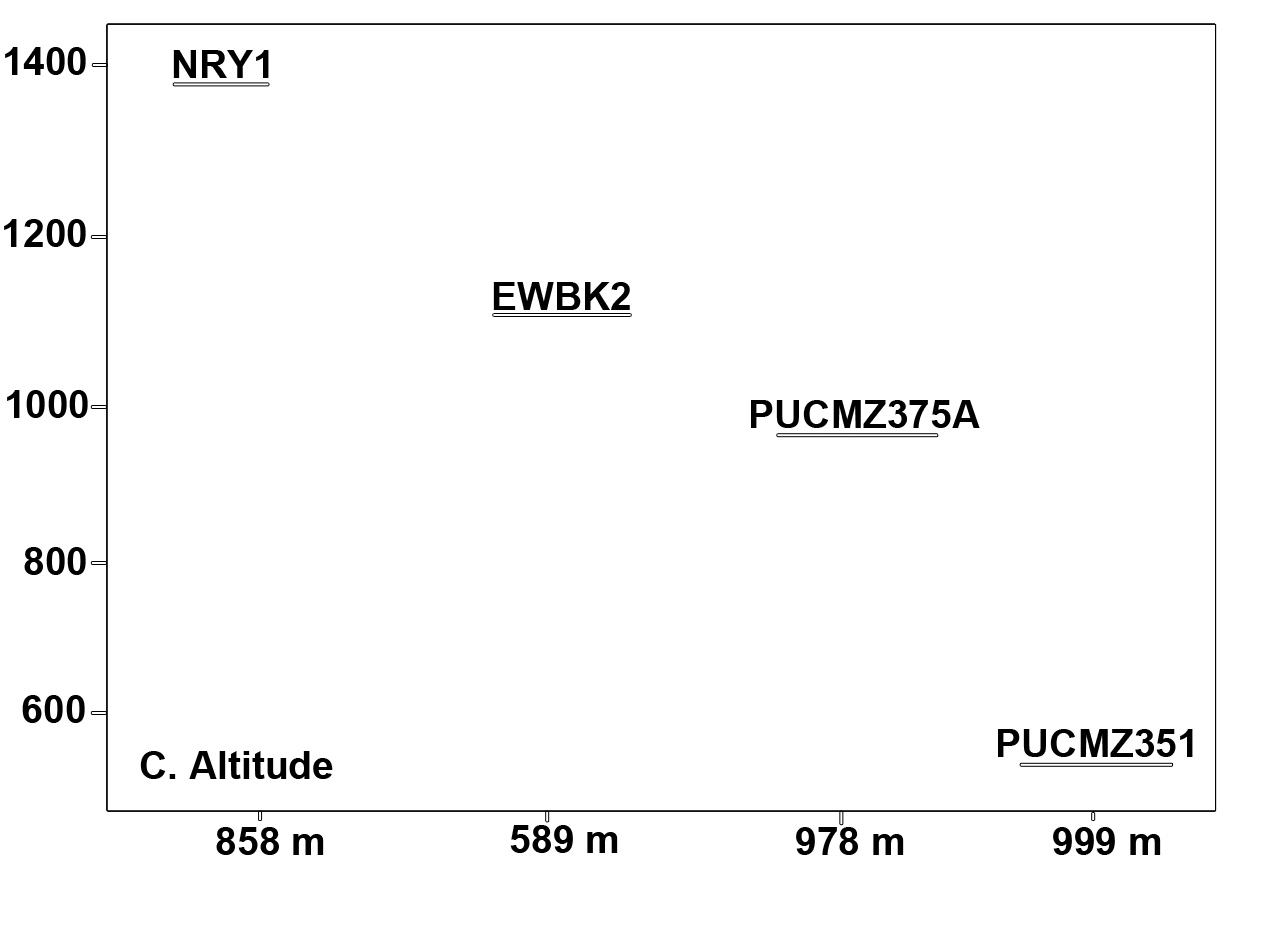
Fig. 1. Alpha diversity measurements of the Metagenome among the earthworm samples (EWBK2-Amynthas alexandri, PUCZM375A-Metaphire houletti,PUCZM351-Eutyphoeus sp, NRY1-Eutyphoeus gigas) around Aizawl city, Mizoram during 2017.
Box plot displaying the diversity difference between (A) pH; (B) Habitat; (C) Altitudinal variations.
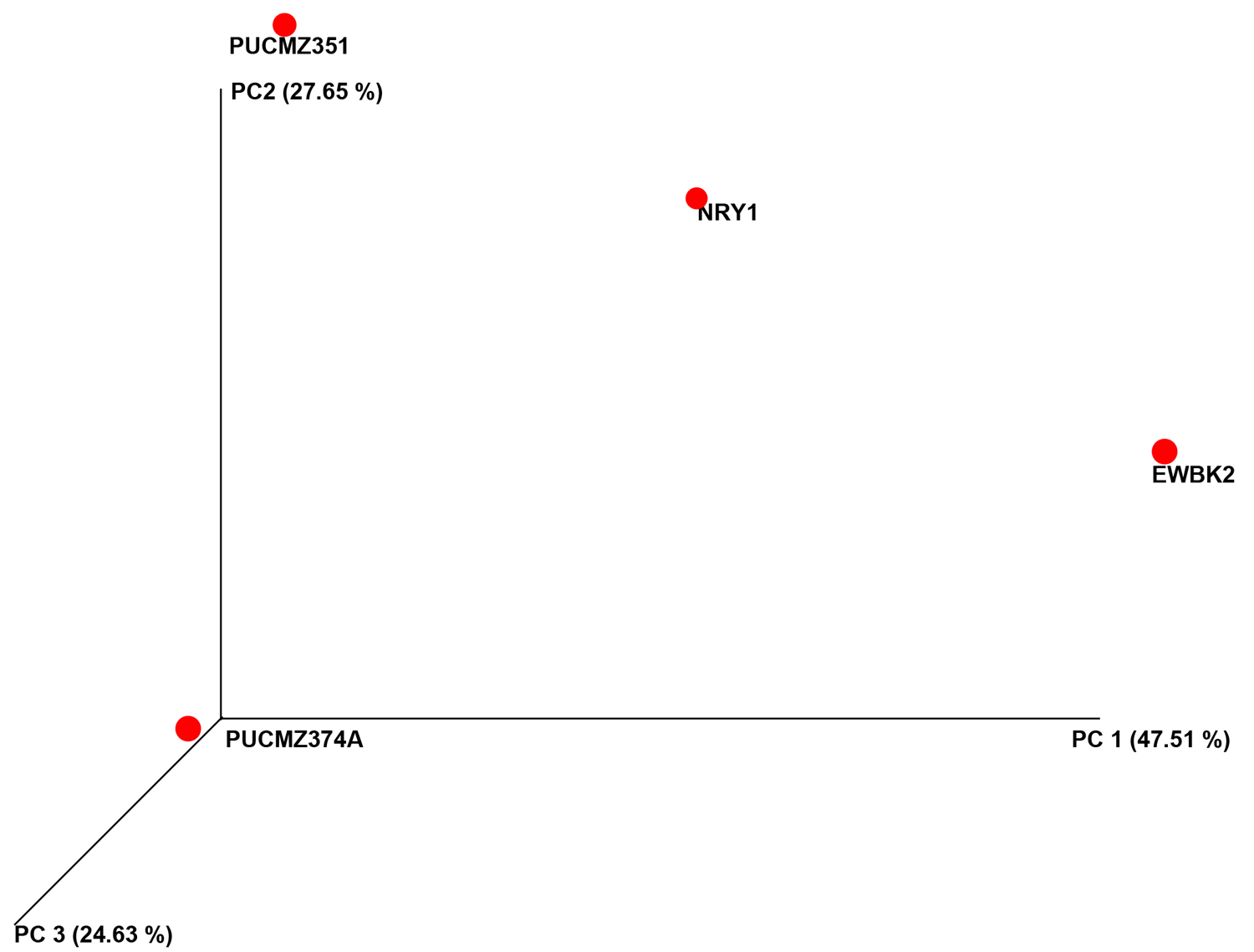
Fig. 2. Beta diversity analysis of the gut microbiota of different earthworm. EWBK2, Amynthas alexandri PUCZM375A, Metaphire houlleti; PUCZM351, Eutyphoeus sp.NRY1, Eutyphoeus gigas.
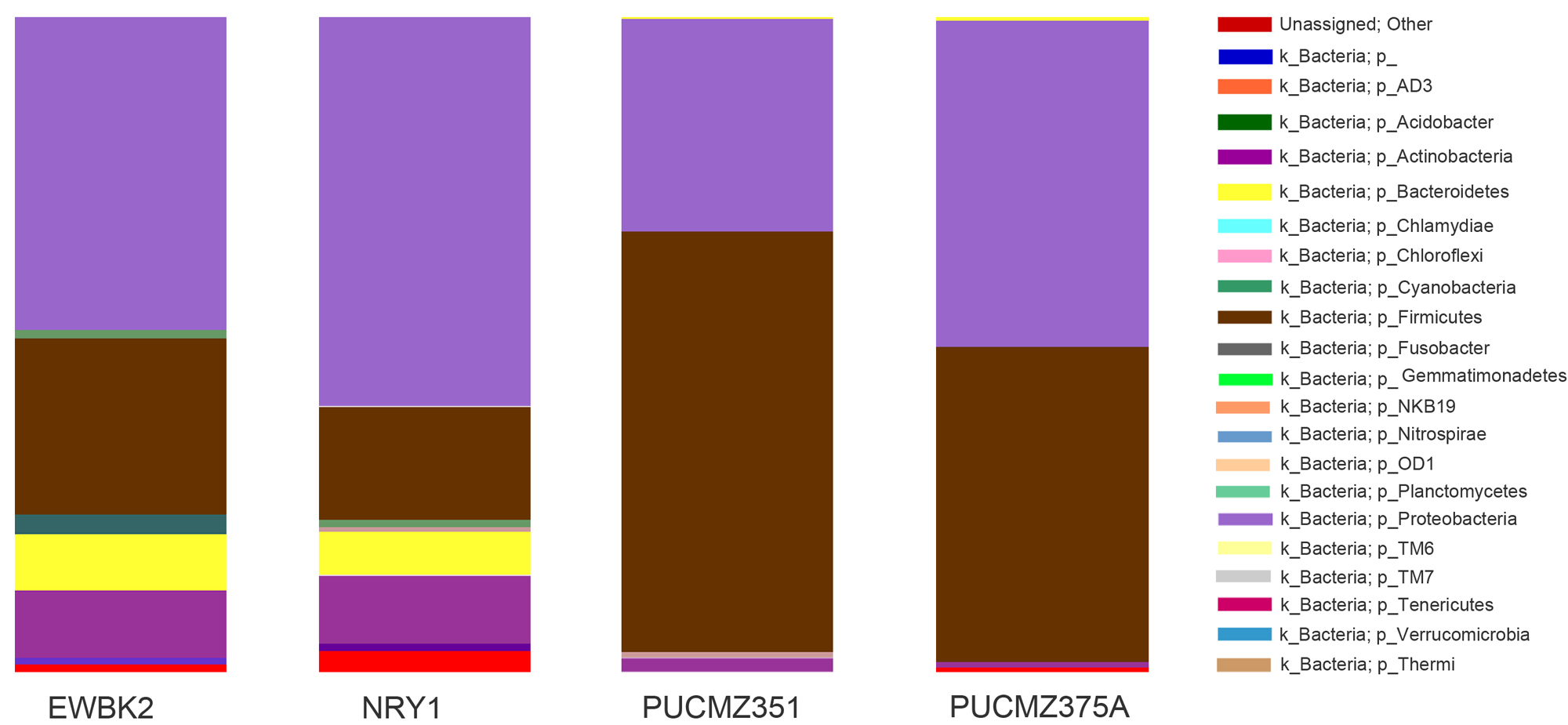
Fig. 3. Taxa distributions at phylum level of individual earthworm species.
EWBK2, Amynthas alexandri; PUCZM375A, Metaphire houlleti; PUCZM351, Eutyphoeus sp. NRY1, Eutyphoeus gigas.
Bacterial community profile
16S rRNA gene amplicon sequencing yielded more than 20 bacterial phyla in the complete dataset (Fig.3). Major bacterial phyla identified were Proteobacteria (47.1%), Firmicutes (38.9%), Actinobacteria (6.3%), Bacteroidetes (3.6%) and Cyanobacteria (1.1%). The other phyla include Chlamydiae, Chloroflexi, Fusobacter, Verrucomicrobia, Fusobacteria, Planctomycetes, Chloroflexi, Fusobacteria, Gemmatimonadetes, NKB19, Nitrospirae, OD1, Planctomycetes, TM6, TM7, Tenericutes, Verrucomicrobia and Thermi. Proteobacteria is dominant in EWBK2, NRY1 and PUZ375A but Firmicutes dominate in PUC351. Bacteroidetes is comparatively low abundant in both habitats. Other phyla such as Actinobacteria, Chlamydiae, Chloroflexi, Fusobacter, Verrucomicrobia, Fusobacteria, Planctomycetes, Chloroflexi, Fusobacteria, Gemmatimonadetes are prominent in home garden habitat compare to forest type. The distributions of bacterial family level were Planococcaceae (27.4%), Enterobacteriaceae (4.1%), Staphylococcaceae (3.5%), Aeromonadaceae (5.6%), Bacillaceae (4.4%), Comamonadaceae (2.4%), Oxalobacteraceae (1.2%), Caulobacteraceae (1.2%) and Enterococcaceae (1.1%). Distribution of bacterial genera were Lysinibacillus (26.9%), Acinetobacter (21.2%), Pseudomonas (4.7%), Bacillus (3.8%), Staphylococcus (3.5%), Stenotrophomonas (1.1%) and Ralstonia (1%).
It was observed that phylum Proteobacteria and Firmicutes are the most abundant in both epigeic and anecic earthworms contributing more than 86% of the total microbial community in both followed by Actinobacteria (Supplementary Fig. 1). Home garden revealed the richer diversity as compared to forest habitat, which might be due to the availability of a variety of food.
Metagenomics analyses of the earthworm gut microbiome: Metabolic potential
Extensive in-silico analysis using the PICRUSt provides the metabolic composition of the earthworm gut microflora. The present work predicts a large number of functional genes involved in various biochemical cycles. It was observed that metagenomic analysis of four species of earthworm gut microbiome revealed the major metabolic substances such as alanine, aspartate and glutamate, amino sugar and nucleotide sugar, arginine and proline, benzoate, butanoate, cysteine and methionine, glycine, serine and threonine, glyoxylate and dicarboxylate, lysine, methane, nitrogen, propanoate, and pyruvate based on the KEGG pathway analysis. Our result revealed the evidence for enrichment of pathways related to nitrate reductase which involved in the nitrate to nitrite. Furthermore, two enzymes such as cytochrome c-type protein and glucosamine-6-phosphate deaminase were identified which participate in the conversion of nitrite to nitric oxide (Fig. 4).
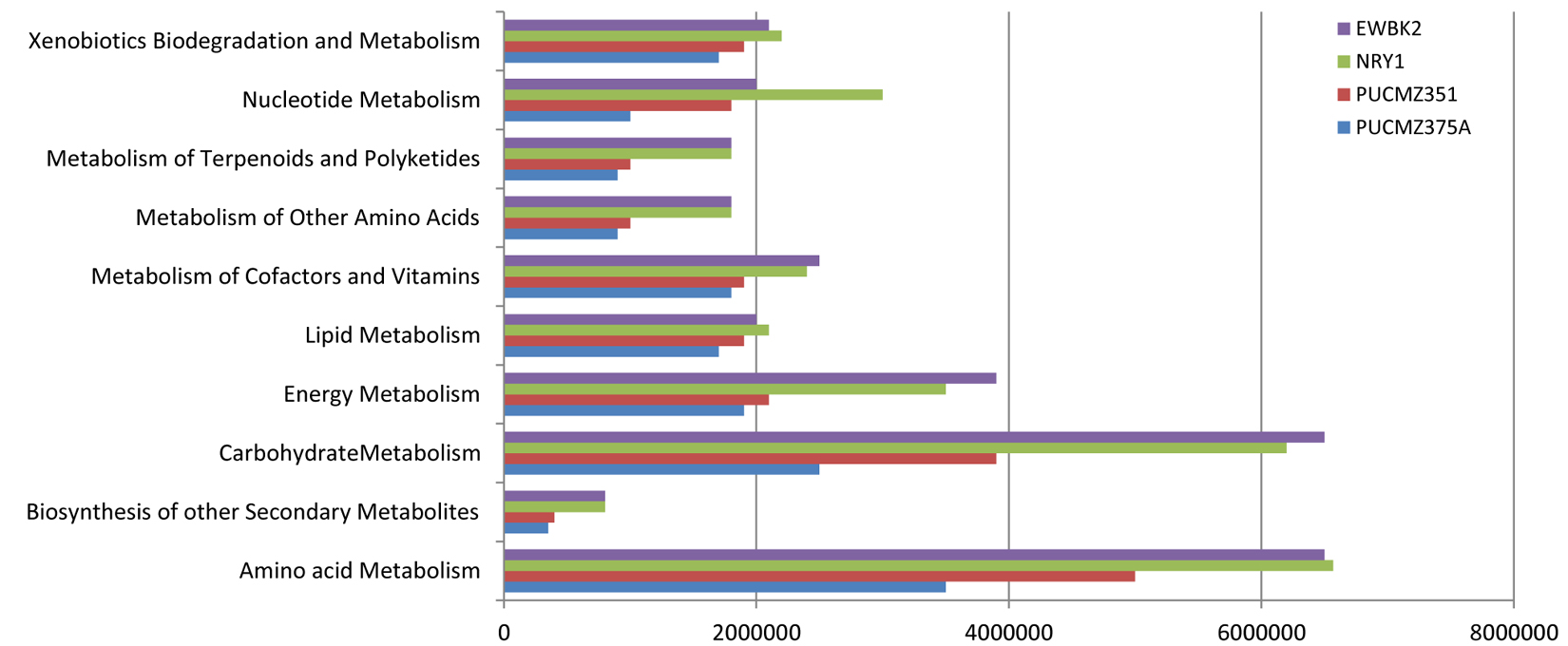
Fig. 4. Predicted metabolic profiles of the earthworm gut microbiome.
EWBK2, Amynthas alexandri; PUCZM375A, Metaphire houlleti; PUCZM351, Eutyphoeus sp.; NRY1, Eutyphoeus gigas.
The present investigation of the earthworm gut bacterial community of Mizoram, northeast India,is the first systematic approach in entire northeast India which emphasizes the gut-bacterial communities of the hilly mountainous biodiversity-rich region.
Earthworm gut contains a unique anaerobic environment and the microbiome is adapted to perform functions including decomposition of nutrients, production of greenhouse gases etc5,8. Factors such as vegetation, pH and altitude play role in the richness and diversity of earthworm species (Fig. 1).The degrees of Alpha diversity in the present study were comparable with other studies investigating the earthworm gut microbiota40. The significant alterations in the observed OTUs and the richness (Chao1 index) suggest that differences between the bacterial communities are mainly compelled by the low abundance OTUs, which is supported by the UniFrac analysis. Our finding revealed a reduction in microbial diversity in the epigeic species. The low bacterial diversity in surface dwellers might be due to the presence of diverse digestive enzymes which allows them to digest microorganisms as well as plant debris25,41. Quantitative changes were noted for the occurrence of bacterial taxa among the samples which could be due to the differences in the feeding substrates. Different substrate components are known to lead to a different microbial community structure42. Deciphering the microbial community would be useful in understanding the ecological function of these earthworms43.
The earthworm gut is inhabited by diverse bacterial communities18-20,39. The present study reveals complex bacterial communities present in the four earthworm samples collected from different places. We found that 10 bacterial genus Bradyrhizobium, Methylobacterium, Mesorhizobium, Rhizobium, Sinorhizobium, Rhodopseudomonas, Oligotropha, Pseudomonas and Mycobacterium were involved in the process of N2O emissions. A similar finding was reported8 from the study on earthworms of Brazil.
Another abundant genus was identified as Streptomyces. Members under this genus were capable of producing cellulase and thereby helping the host to degrade plant materials43. Another dominant genus Pseudomonas possess genes involves in terpene metabolism and thereby participate in plant litter decomposition45. One of the stimulating observations was the high abundance of the bacterial genus Lysinibacillus. Members under this genus participate in polyethylene degradation46-50 and xylan biodegradation could be an appropriate candidate for forest-based waste degradation51. Thus, this earthworm species can be further explored for forest waste biomass vermicomposting.
Stenotrophomonas is a common soil microbe, it plays a central role in nitrogen fixation as well as cellulolytic activity52-54. Another dominant genus, Acinetobacter is a common intestinal species found to be involved in the decomposition of catechin (plant secondary metabolite)55. The genus Acinetobacter is enhanced during fermentation. The abundance of this genus Acetobacter was possibly due to the anoxic environment and high organic substrates existing in the gut stimulate of the earthworm species56-57. We also identified the genus Methylococcus, belonging to the bacterial group Methylotrophs which capable of utilizing reduced one-carbon compounds, methanol or methane as the carbon source, and multi-carbon compounds such as dimethyl ether and dimethylamine58.
Due to the variation in location as well as feeding habits, different groups of earthworms harbour different bacterial communities. Our result indicates that major differences observed within bacterial phyla Firmicutes, Actinobacteria and Bacteroidetes. Firmicutes have significantly decreased in surface dwellers (epigeic) species but increased in deep soil inhabitants (anecic worms). Actinobacteria and Bacteroidetes were increased in sub-surface earthworms (endogeic) compared to anecic species59. Previous studies reported that the gut metagenome of the anecic earthworms was dominated by the Beta proteobacteria and Gammaproteobacteria59. The present study is also in line with this report in having proteobacteria as the dominant phylum. However, Firmicutes60 is also a dominant phylum in our study, which is not following the temperate region. This may be attributed to geographical differences in the two studies.
The metabolic activity of the earthworm gut microbial community of the present study showed an abundant representation of genes which represent pathways related to metabolism including the metabolism of amino acid, carbohydrate, energy, cofactor and degradation of xenobiotics. The abundance of these metabolic modules may be due to the feeding habitat of the analyzed earthworms. The metabolic profile of the species EWBK2 and NRY1 were similar compared to other species. This probably is due to the forest environment which allows them to ingest a large amount of plant material which is normally rich in cellulose, hemicellulose and soluble carbohydrates61. In conformity with the taxonomic characterization, we found the representative genes involved in denitrification, methanogenesis. This is an important function played by the gut bacterial community which helps in recycling nitrogen waste of the host62.
Illumina sequencing reveals the diversity of bacterial communities in earthworm gut micro-biota from Indo-Myanmar Biodiversity Hotspot and predicts their imputed metabolic profiles. This bacterial diversity can be attributed to differences in the environmental condition such as pH, altitude, and type of forest. Overall, this study gives insight into the gut microbial composition and their putative functional roles in the gut of tropical hilly earthworms. Further studies on the functional roles of these microbes will enlighten the ecosystem functioning and geo-biochemical cycles.
ACKNOWLEDGMENTS
The authors express their gratitude to the Department of Biotechnology (DBT) for the Earthworm project (DBT/PR11849/NER/9509/2014) as well as Bioinformatics Infrastructure Facility (BT/BI/12/060/2012 (NERBIFMUA), Mizoram University and Advance Level Biotech-Hub, Pachhunga University College, Aizawl, India. The authors thank Dr. Lalthansangi Fanai, Department of English, Government Aizawl College, India for language editing.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
AUTHORS’ CONTRIBUTION
All authors listed have made a substantial contribution to the work and approved it for publication.
FUNDING
This research was sponsored by Department of Biotechnology, Govt. of India, New Delhi.
ETHICS STATEMENT
This work does not contain any studies with human participants or animals performed by any of the authors.
AVAILABILITY OF DATA
The sequences obtained from NGS were submitted to NCBI which are available under Bio Project ID PRJNA376467.
- Jones CG, John HL, Moshe S. Organisms as ecosystem engineers. Oikos.1994;69(3):373-386.
Crossref - Edwards CA. Earthworm Ecology. 2nd Ed. CRC Press, LLC, 2000 N.W. Corporate Blvd., Boca Raton, Florida 33431. 2004;1-417.
- Owa SO, Olowoparja SB, Aladesida A,Dedeke GA. Enteric bacteria and fungi of the Eudrilid earthworm Libyodrilus violaceus. Afr J Agri Res. 2013;8(17):1760-1766.
Crossref - Edwards CA, Fletcher KE. Interactions between earthworms and microorganisms in organic matter breakdown. Agri. Ecosyst. Environ. 1988;24:235-247.
Crossref - Horn MA, Schramm A, Drake HL. The earthworm gut: An ideal habitat for ingested N2O-producing microorganisms. Appl Environ Microbiol. 2003;69:1662-1669.
Crossref - Thakuria D, Schmidt O, Finan D. Gut wall bacteria of earthworms: a natural selection process. The ISME J. 2010;4:357-366.
Crossref - Wust PK, Horn MA, Drake H.L. Clostridiaceae and Enterobacteriaceae as active fermenters in earthworm gut content. ISME J. 2011;5:92-106.
Crossref - Peter SDJ, Siu MT, Marcus AH, Harold LD. Emission of nitrous oxide and dinitrogen by diverse earthworm families from Brazil and resolution of associated denitrifying and nitrate-dissimilating taxa. FEMS Microbial Ecol. 2013;83(2):375-391.
Crossref - Hornor SG, Mitchell MJ. Effect of the earthworm, Eisenia foetida (Oligochaeta), on fluxes of volatile carbon and sulfur compounds from sewage sludge. Soil Biol Biochem. 1981;13(5):367-372.
Crossref - Drake HL, Horn MA. As the worm turns: the earthworm gut as a transient habitat for soil microbial biomes. Ann Rev Microbiol. 2007;61:169-189.
Crossref - Joshi NV, Kelkar BV. The role of earthworms in soil fertility. Ind J Agri Sc. 1952;22:189-196.
- Curry JP, Schmidt O. The feeding ecology of earthworms–a review. Pedobiologia. 2007;50(6):463-477.
Crossref - Xing R, Gao QB, Zhang FQ. Bacterial community in cold and alkaline environments of Hoh Xil basin in Qinghai–Tibet Plateau and isolation of potential sources of microbiota. Ann Microbiol. 2019;69(5):567-576.
Crossref - Han J, Sun L, Dong X, et al. Characterization of a novel plant growth-promoting bacteria strain Delftiatsuruhatensis HR4 both as a diazotroph and a potential biocontrol agent against various plant pathogens. Syst Appl Microbiol. 2005;28(1):66-76.
Crossref - Sunish KR, Ayyadurai N, Pandiaraja P, et al. Characterization of antifungal metabolite produced by a new strain Pseudomonas aeruginosa PuPa3 that exhibits broad-spectrum antifungal activity and biofertility traits. J Appl Microbiol. 2005;98:145-154.
Crossref - Ihssen J, Horn MA, Matthies C, Gobner A, Schramm A, Drake HL. N2O-Producing Microorganisms in the Gut of the Earthworm Aporrectodeacaliginosa Are Indicative of Ingested Soil Bacteria. Appl Environ Microbiol. 2003:69(3):1655-1661.
Crossref - Brito-Vega H, Espinosa-Victoria D. Bacterial Diversity in the Digestive Tract of Earthworms (Oligochaeta). J Biol Sci. 2009;9(3):192-199.
Crossref - Govindarajan B, Prabaharan V. Gut micro-flora of earthworms: A review. Amer J Biol Pharma Res. 2014;1(3): 125-130.
- Khyade VB. Bacterial diversity in the alimentary canal of earthworms. JBacteriol Mycol Open Access. 2018;6(3):183-185.
Crossref - Sapkota R, Santos S, Farias P, Krogh PH, Winding A. Insights into the earthworm gut multi-kingdom microbial communities. Sci Total Environ. 2020;727:138301
Crossref - Singh A, Singh DP, Tiwari R, et al. Taxonomic and functional annotation of gut bacterial communities of Eisenia foetidaand Perionyx excavatus. Microbiol Res. 2015;175:48-56.
Crossref - Yadav S. Metagenomics Explorations of Earthworm Gut Micro-Biome to Detoxify NPS in Soil System. Int J Rec Sci Res. 2016;7(4):10113-10116.
- Samanta TT, Das A. Isolation, identification, and characterization of gut microflora of Perionyx excavatus collected from Midnapore, West Bengal. J Basic Microbiol. 2016;56:286-293.
Crossref - Biswas S, Lahiri P, Das S. Isolation of predominant bacterium from gut of earthworm Lampitomauritii for effective use in soil fertility. Curr Sci. 2014;107(1):105-109.
- Gomez-Brandon M, Aira M,Lores M, Dominguez J. Epigeic earthworms exert a bottleneck effect on microbial communities through gut associated processes. PloS One. 2011;6(9):e24786.
Crossref - Medina-Sauza RM, Alvarez-Jimenez M, Delhal A, et al. Earthworms Building Up Soil Microbiota, a Review. Front Environ Sci. 2019;7:81.
Crossref - Stephenson J. The Fauna of British India including Ceylon and Burma. Oligochaeta.1923; xxiv + 518 pp. London: Taylor and Francis.
- Gates GE. Burmese earthworms. An introduction to the systematics and biology of megadrile oligochaetes with special reference to Southeast Asia. Trans Amer Phil Soc. 1972;62(7):1-326.
Crossref - Julka JM. The Fauna of India and the adjacent countries. Megadrile Oligochaeta (Earthworms). Haplotaxida: Lumbricina: Megascolecidea. 1988. xiv+400 pp.Zoological Survey of India, Calcutta.
- Ravindran B, Contreras-Ramos SM, Sekaran G. Changes in earthworm gut associated enzymes and microbial diversity on the treatment of fermented tannery waste using epigeic earthworm Eudriluseugeniae. Ecol Engineer. 2015;74:394-401.
Crossref - Hussain N, Singh A, Saha S, Kumar MVS, Bhattacharyya P, Bhattacharya SS. Excellent N-fixing and P-solubilizing traits in earthworm gut-isolated bacteria: A vermicompost based assessment with vegetable market waste and rice straw feed mixtures. Biores Tech. 2016;222:165–174.
Crossref - Caporaso JG, Bittinger K, Bushman FD, DeSantis TZ, Andersen GL, Knight R. PyNAST: A flexible tool for aligning sequences to a template alignment. Bioinformatics. 2010;26:266-267.
Crossref - Caporaso JG, Kuczynski J, Stombaugh J, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335-336.
Crossref - Jones RT, Robeson MS, Lauber CL, Hamady M, Knight R, Fierer N. A comprehensive survey of soil acidobacterial diversity using pyrosequencing and clone library analyses. The ISME J. 2009;3(4):442-453.
Crossref - Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27:2194-2200.
Crossref - Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460-2461.
Crossref - De Santis TZ, Hugenholtz P, Larsen N, et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. 2006;72(7):5069-5072.
Crossref - De Mandal S, Chatterjee R, Kumar NS. Dominant bacterial phyla in caves and their predicted functional roles in C and N cycle. BMC Microbiol. 2017;17:90.
Crossref - Langille MGI, Zaneveld J, Caporaso JG, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. 2013;31:814-821.
Crossref - Liu D, Lian B, Wu C, Guo P. A comparative study of gut microbiota profiles of earthworms fed in three different substrates. Symbiosis. 2018;74:21-29.
Crossref - Zhang BG, Li GT, Shen TS, Wang JK, Sun Z. Changes in microbial biomass C, N, and P and enzyme activities in soil incubated with the earthworms Metaphire guillelmi or Eisenia fetida. Soil Biol Biochem. 2000;32:2055-2062.
Crossref - Engel P, Moran NA. The gut microbiota of insects – diversity in structure and function. FEMS Microbiol Rev. 2013;37:699-735.
Crossref - Drake HL, Schramm A, Horn MA. Earthworm gut microbial biomes: their importance to soil microorganisms, denitrification, and the terrestrial production of the greenhouse gas N2O. In Intestinal microorganisms of Termites and Other Invertebrates, 2005; pp. 65-87. Springer, Berlin, Heidelberg.
Crossref - Prasad P, Singh T, Bedi S. Characterization of the cellulolytic enzyme produced by Streptomyces griseorubens (Accession No. AB184139) isolated from Indian soil. J King Saudi Univ Sc. 2013;25:245-250.
Crossref - Campos-Garcia J. Metabolism of acyclic terpenes by pseudomonas. In: Pseudomonas. 2010;235-253.
Crossref - Koutny M, Lemaire J, Delort AM. Biodegradation of polyethylene films with prooxidant additives. Chemosphere. 2006;64(8):1243-1252.
Crossref - Arutchelvi J, Sudhakar M, Arkatkar A, Doble M, Bhaduri S, Uppara PV. Biodegradation of polyethylene and polypropylene. Ind J Biotechnol. 2008;7(1):9-22.
- Bhardwaj H, Gupta R, Tiwari A. Communities of microbial enzymes associated with biodegradation of plastics. J. Pol. Environ. 2012;21(2):575-579.
Crossref - Restrepo-Florez JM, Bassi A, Thompson MR. Microbial degradation and deterioration of polyethylene-a review. Int Biodeterior Biodegrad. 2014;88:83-90.
Crossref - Grover A, Gupta A, Chandra S, Kumari A, Khurana SM. Polythene and environment. Int J Environ Sc. 2015;5(6):1091-1105.
- Lee CS, Jung YT, Park S,Oh TK, Yoon JH. Lysinibacillus xylanilyticus sp. nov., a xylandegrading bacterium isolated from forest humus. Int J Sys Evol Microbiol. 2010;60:281-286.
Crossref - Morales-Jimenez J, Zuniga G, Ramirez-Saad HC. Gut-Associated Bacteria Throughout the Life Cycle of the Bark Beetle Dendroctonus rhizophagus Thomas and Bright (Curculionidae: Scolytinae) and their cellulolytic activities. Microbial Ecol. 2012;64:268-278.
Crossref - Morales-Jimenez J, Zuniga G, Villa-Tanaca L. Bacterial community and nitrogen fixation in the red turpentine beetle, Dendroctonus valens LeConte (Coleoptera: Curculionidae: Scolytinae). Microbial Ecol. 2009;58:879-891.
Crossref - Adams AS, Aylward FO, Adams SM, et al. Mountain pine beetles colonizing historical and naive host trees are associated with a bacterial community highly enriched in genes contributing to terpene metabolism. Appl Environ Microbiol. 2013;79:3468-3475.
Crossref - Arunachalam PS, Yang SH, Damodharan K, Suh JW. Genetic and functional characterization of culturable plant-beneficial actinobacteria associated with yam rhizosphere. J Basic Microbiol. 2013;53: 985-995.
Crossref - Horn MA, Drake HL, Schramm A. Nitrous oxide reductase genes (nosZ) of denitrifying microbial populations in soil and the earthworm gut are phylogenetically similar. Appl Environ Microbiol. 2006;72(2):1019-1026.
Crossref - Horn JL, Danuta PJ, Hamer ML. The leaf-litter earthworm fauna (Annelida: Oligochaeta) of forests in Limpopo Province, South Africa: diversity, communities and conservation. Afr Zool. 2007;42(2):172-179.
Crossref - Anthony C. The biochemistry of methylotrophic micro-organisms. Sc Prog. 1975;62:167-206.
- Aira M, Bybee S, Perez-Losada M, Dominguez J. Feeding on microbiomes: effects of detritivory on the taxonomic and phylogenetic bacterial composition of animal manures. FEMS Microbiol Ecol. 2015;91(11).
Crossref - Singleton DR, Hendrix PF, Coleman DC, Whitman WB. Identification of uncultured bacteria tightly associated with the intestine of the earthworm Lumbricusrubellus (Lumbricidae; Oligochaeta). Soil Biol Biochem. 2003;35(12):1547-1555.
Crossref - Bahrndorff S, De Jonge N, Hansen JK, et al. Diversity and metabolic potential of the microbiota associated with a soil arthropod. Scient Rep. 2018;8(1):2491.
Crossref - Warnecke F, Luginbuhl P, Ivanova N, et al. Metagenomic and functional analysis of hindgut microbiota of a wood-feeding higher termite. Nature. 2007;450:560-565.
Crossref
© The Author(s) 2020. Open Access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License which permits unrestricted use, sharing, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.