


ISSN: 0973-7510
E-ISSN: 2581-690X
Chronic obstructive pulmonary disease (COPD) and tuberculosis (TB) are major global health burdens that often coexist, potentially amplifying disease severity through shared inflammatory and immune dysregulation mechanisms. This study aimed to elucidate the molecular interplay between these diseases, providing insights that could inform targeted therapies. Through integrative analyses, including differential gene expression, pathway enrichment, protein-protein interaction (PPI) networks, and tissue-specific expression profiling, we identified 38 genes central to the shared pathophysiology of COPD and TB. Key genes such as RELA, RELB, and PLK3 were consistently upregulated, underscoring their roles in chronic inflammation and immune signaling, while downregulated genes like IKBIP and MS4A6A highlighted suppressed immune pathways, potentially linked to immune evasion strategies. Pathway enrichment revealed significant involvement of NF-κB signaling, cytokine-mediated pathways, and metabolic adaptations, emphasizing their relevance in maintaining inflammation and pathogen persistence. PPI analysis identified regulatory hubs such as STAT1 and guanylate-binding proteins (GBP5, GBP1), highlighting their potential as therapeutic targets. Tissue-specific expression pinpointed the lungs and whole blood as critical sites of transcriptional activity, while single-cell RNA sequencing localized dysregulated genes to alveolar macrophages and epithelial cells, revealing their cell-specific roles in disease progression. These findings provide a foundation for understanding the shared molecular mechanisms underpinning COPD and TB, emphasizing immune signaling and metabolic regulation as potential therapeutic avenues. Further experimental validation and multi-omics studies are essential to translate these insights into clinical applications, addressing the complex interplay of these coexisting diseases.
COPD, Tuberculosis, Molecular Signatures, DEG, Systems Biology, PPI, Transcriptomics
Chronic Obstructive Pulmonary Disease (COPD) and tuberculosis (TB) are significant global health challenges, each contributing substantially to morbidity and mortality worldwide. Chronic Obstructive Pulmonary Disease (COPD) is caused by extensive exposure to noxious particles or gases, the most prevalent factor being tobacco smoking. Its clinical manifestations include persistent respiratory symptoms as well as airflow impairment.1,2 As per the statistics provided by the World Health Organization (WHO), this disease was the third most prominent cause of death across the globe in the year 2019, being the underlying cause of 3.23 million deaths.3 Conversely, tuberculosis, caused by Mycobacterium tuberculosis (Mtb), remains the leading cause of death from infectious diseases, with 10 million new cases and 1.4 million deaths recorded in 2020.4,5 The coexistence of COPD and TB not only exacerbates the clinical burden of each disease but also presents unique challenges due to overlapping mechanisms of immune dysregulation, chronic inflammation, and metabolic alterations.6,7
Chronic Obstructive Pulmonary Disease (COPD) is caused primarily by the smoking of tobacco as it triggers an inflammatory response in the airways is accompanied by a significant probability of demonstrable tissue damage, as seen in lung tuberculosis. Tuberculosis (TB), on the contrary, displays a strong immune response that may even encapsulate the Mycobacterium tuberculosis (Mtb), which in this case is seen leading to the growth of granulomas that help in delimiting the spread of the infection to other tissues.8-10 Chronic inflammation is a pivotal factor in the progression of both conditions, and their simultaneous occurrence can initiate a detrimental cycle of immune activation, tissue damage, and exacerbation of clinical symptoms. There is also a higher incidence of TB in cases where an individual is suffering from COPD, but the presence of an underlying TB can worsen the TB’s clinical picture and disorders like COPD.11-13 Notwithstanding the clinical relevance of co-infection with COPD and TB, the underlying molecular mechanisms facilitating their interaction remain inadequately elucidated. Most studies have focused on the individual pathophysiology of COPD and TB, leaving a gap in our understanding of their shared molecular and transcriptional signatures.14,15 Although the overlapping inflammatory and immune mediators implicated in these diseases are widely acknowledged, the exact regulatory mechanisms and pathways involved have yet to be thoroughly delineated. It is imperative to bridge this divide in order to identify potential therapeutic targets that can effectively address the distinctive challenges posed by co-infection.
The possibilities provided by the simultaneous interplay of systems biology and high throughput techniques allow for the combined examination of gene transcription, combinatorial proteins, and pathways enrichment. These methods open up new ways to study common aberrations of gene transcription, immune system pathways, and metabolic changes that characterize COPD and TB’s co-pathophysiology. In particular, transcriptomic profiling, pathway enrichment analysis, and protein-protein interaction (PPI) network analysis have proven invaluable in identifying key regulatory genes and pathways that drive chronic inflammatory diseases. These integrated studies are important for discovering molecular mechanisms for specific targeted treatment strategies.16-19 To achieve the goal of the present study, that is, to unravel the common molecular basis of COPD and TB, systems biology approaches were employed. In this case, we focus on differentially expressed genes across stimulated and unstimulated conditions to find common transcriptional patterns between the two diseases. We utilize pathway enrichment and PPI network analyses to elucidate the immune signaling pathways and the metabolic changes underlying the chronic inflammation of COPD and TB. Moreover, tissue and cell type-specific genetics studies reveal where the dysregulated genes are situated within organs and broaden the understanding of processes at both local and systemic involvement.
Identifying shared genes would help define the impact of immune dysregulation and inflammation for tuberculosis and chronic obstructive pulmonary disease comorbidity. The genes show the common pathways that lead to chronic inflammation, whereas the different genes show the impairment of immune signals. Tissue-specific and single-cell analyses further contextualize these findings, identifying critical roles for alveolar macrophages and epithelial cells in localized immune responses and systemic implications in whole blood. By elucidating shared transcriptional signatures and molecular pathways, this study provides a foundation for developing therapeutic interventions that target common mechanisms of COPD and TB co-infection. The workflow of the study is given in Figure 1. These findings contribute to a deeper understanding of the pathophysiology of these diseases and lay the groundwork for future research integrating multi-omics and longitudinal approaches to validate and expand upon these insights.
Figure 1. Study workflow showcasing the key steps and methodologies, from data collection through analysis to the final results
Dataset acquisition and experimental design
The gene expression data for this study was obtained from publicly available repositories hosted on the Gene Expression Omnibus (GEO)20 database under accession numbers GSE3460821 and GSE56768.22 For GSE34608, microarray experiments were carried out using Agilent-014850 Whole Human Genome Microarray 4x44K GPL6480 platform. This platform has a clear advantage when conducting genomic-wide expression profiling, as mRNA expression for many genes can be measured. It uses the two-color hybridization system to obtain accurate and reproducible gene expression measurements. Likewise, the dataset GSE56768 applied the Affymetrix Human Genome U133 Plus 2.0 Array (GPL570). This platform includes over 47,000 most of human transcriptomes. It is widely used for gene expression studies due to its high reliability and reproducibility. Both platforms ensure reliable and reproducible results, crucial for identifying differentially expressed genes in tuberculosis, COPD, and control samples.
Sample grouping and experimental design
The gene expression datasets for tuberculosis (GSE34608) and chronic obstructive pulmonary disease (GSE56768) were curated and grouped according to experimental conditions for differential expression analysis. Two groups that were recruited included 8 samples from tuberculosis patients and 18 healthy controls for tuberculosis. In cases of COPD, a total of 125 samples were subdivided into four distinct subsets including 40 samples from “COPD Stimulated (CD)”, 47 samples from “COPD Unstimulated”, 20 samples from “Smoker Stimulated”, and 18 samples from Smoker Unstimulated. These groupings allowed thorough analysis of gene expression variations under many disease conditions and the immune response.
Data preprocessing, normalization, and statistical analysis
The expression datasets downloaded from GEO (Raw datasets: GSE34608, GSE56768) were pre-processed for any further analysis. After determining their distribution through the quantile statistics, the values provided were first transformed to log2, indicating that the variance across datasets needed to be equalized.23 To only keep the biologically relevant data, probes with missing expression data and those that did not map to genes or control probes were left out. Quantile normalization was performed to eliminate technical variation and ensure comparability across samples, minimizing batch effects arising from differences in experimental conditions or platforms.24
Differential expression analysis was conducted using the limma package in R, designed for microarray datasets. A design matrix was constructed to represent group labels, and contrasts of interest were specified for comparisons. Linear models were fitted using the lmFit function, and moderated t-statistics were calculated via the Bayes method, which improves robustness, particularly for smaller sample sizes. Genes with an adjusted p-value of less than 0.05 (Benjamini-Hochberg correction) and a log2 fold-change exceeding ±1.5 were deemed significantly differentially expressed.25 Visualizations such as histograms of p-value distributions, Q-Q plots, volcano plots, and mean-difference plots were generated to assess data quality and highlight significant findings. Dimensionality reduction using UMAP facilitated visualization of clustering patterns among sample groups, while a Venn diagram illustrated overlapping differentially expressed genes across groups,26 providing insights into shared molecular mechanisms.
Identifying common genes
Shared molecular mechanisms between Chronic Obstructive Pulmonary Disease (COPD) and Mycobacterium tuberculosis (Mtb) were investigated by identifying common differentially expressed genes. From the analysis of datasets GSE34608 and GSE56768, 38 genes were found to overlap between the two conditions. These genes represented potential shared regulatory mechanisms, while 392 and 1207 genes were unique to COPD and Mtb, respectively. The identified genes were selected for downstream functional and pathway analyses to explore their roles in disease progression.
Gene ontology and pathway enrichment analysis
To understand the biological processes and pathways common for COPD and Mtb, we performed Gene Ontology (GO) and Pathway enrichment analyses. We utilized different databases like KEGG27 and Reactome28 to conduct GO analyses. We selected relevant biological processes, molecular functions, and cellular components. Adjusted p-values (FDR < 0.05) were used for enrichment analysis, influencing the determination of the relevant significant terms. GSEA was also performed with various gene set libraries, including ChEA 2022 and others Pathway Collection, using the Enricher platform,29 leveraging multiple gene set libraries, including ChEA 2022 and others Pathway Collection, to further contextualize the shared genes within broader regulatory networks and pathways. The outcomes were comprehensively depicted using bar graphs and cluster maps of the vital pathways and their interrelations, ensuring a thorough indication of the functional architecture of the shared gene set.
Protein-protein network interactions
The STRING database30 was employed for protein interaction networks with a minimum interaction score of >0.7 to have a reasonable level of confidence in the relations. The network was visualized and further analyzed using Cytoscape31 to determine the important interactions within the network. Critical functional modules were also highlighted by the MCODE algorithm, which also finds dense clusters of interactions amongst proteins. For more focused study, these clusters were ranked on the basis of their MCODE scoring to determine the critical proteins and pathways that required more detailed study.
Single cell multi-gene expression analysis in lung tissue
In line with the Gene Ontology and Pathway Enrichment Analysis results, the GTEx portal32 was used to carry out a Single-cell multi-gene expression analysis. This particular study aimed to characterize the expression patterns of the elucidated co-regulated genes within lung tissues and whole blood. The method allowed deep explorations of the gene expression patterns at the tissue level and enabled the understanding of the functional context of the genes within the biological system of interest.
The study aims to investigate the transcriptional landscape of both COPD & TB through different experimental scenarios to seek out common molecular signatures. COPD was studied in under-stimulated and unstimulated conditions, including subgroups such as smoker-unstimulated and smoker-stimulated samples. This approach aimed to capture the impact of immune modulation and smoking status on gene expression. In the same way, the TB dataset was analyzed to determine whether there are specific transcriptional changes in the presence of the disease when compared with healthy controls. The primary purpose of this study was to start looking for the common molecular mechanisms or molecular overlaps by identifying the upregulated and downregulated genes between COPD and Tuberculosis. This work used specialized bioinformatics tools like differential expression analysis, Gene Ontology annotation, pathway analysis, protein-protein interaction network, single-cell gene expression profiling, etc., to provide an in-depth transcriptional data analysis and highlight significant genes of interest. This work addresses the need to understand better the molecular relationships between COPD and TB, especially in the case of the 36 common genes found in both conditions. These genes are suggested to be involved in the diseases’ basic immune mechanisms and responses and, therefore should be considered for future work in experimental validation or therapies.
Quality control and normalization of gene expression data
The expression data from COPD (GSE56768) and Tuberculosis (GSE34608) datasets were evaluated for quality and consistency after normalization to ensure robust and reliable analysis. The boxplots (Figure 2A and 2B) demonstrate the distribution of normalized expression values across various sample groups, including COPD unstimulated, COPD stimulated, smoker stimulated, smoker unstimulated, tuberculosis, and healthy controls. These visualizations reveal consistent median expression levels across samples, indicating successful normalization and minimization of technical variability. Further confirmation of data uniformity was obtained through density plots (Figure 2A and 2B), which display overlapping distribution curves for the expression intensities of samples within each dataset. This alignment suggests the absence of batch effects and ensures comparability across experimental conditions. Such preprocessing quality checks were critical to the reliability of the downstream differential expression analysis.
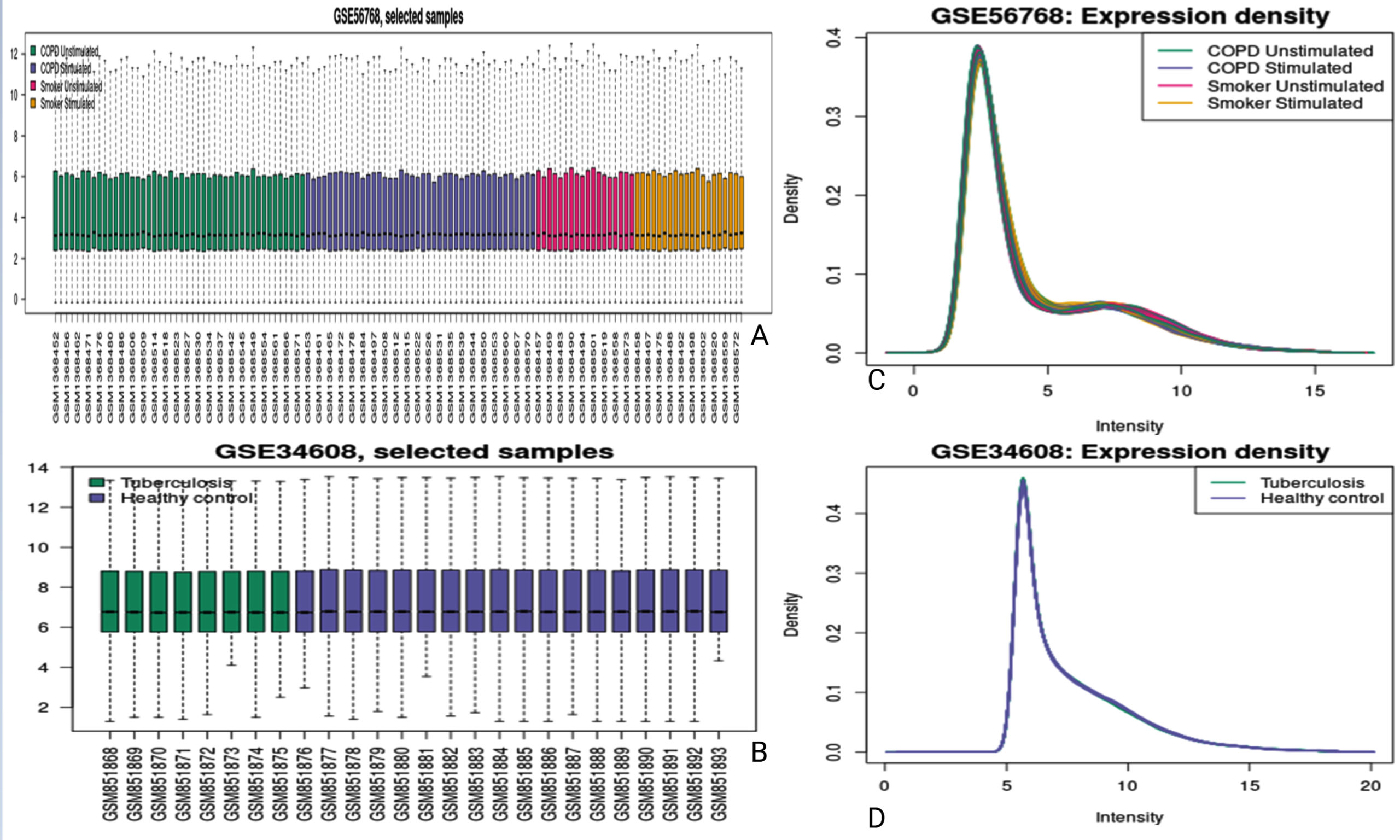
Figure 2. Expression profiles and density distributions for selected samples from the GSE56768 and GSE34608 datasets. Image (A) shows a boxplot of gene expression across four conditions in the GSE56768 dataset: COPD unstimulated, COPD stimulated, smoker unstimulated, and smoker stimulated. Image (B) presents a boxplot comparing gene expression between tuberculosis patients and healthy controls from the GSE34608 dataset. Image (C) displays the density distribution of expression intensities for the conditions shown in image (A), while image (D) shows the corresponding density distribution for the groups in image (B)
Table:
Gene Expression Datasets for Tuberculosis and COPD
GEO Accession |
Sample Size |
Platform |
Tissue |
|---|---|---|---|
GSE34608 |
8 TB + 18 Healthy |
GPL6480 |
Gene and microRNA expression in pulmonary tuberculosis and sarcoidosis |
GSE56768 |
40 COPD Stimulated + 47 COPD Unstimulated + 20 Smoker Stimulated + 18 Smoker Unstimulated |
GPL570 |
Whole blood and isolated blood cell transcriptomics in COPD |
Differentially Expressed Genes (DEG) analysis and common gene identification
The differential expression analysis identified significant alterations in gene expression across the five comparison groups (Table). Each group was analyzed to detect both upregulated and downregulated genes based on predefined criteria (adjusted p-value < 0.05 and log2 fold-change > ±1.5). The analysis revealed varying numbers of DEGs across comparisons. For instance, comparing COPD Unstimulated and COPD Stimulated resulted in 11,222 DEGs, with 6,150 genes upregulated and 5,072 downregulated. Similarly, comparing COPD Stimulated versus Smoker Unstimulated identified 11,129 DEGs, and the Healthy Control versus Tuberculosis group yielded 1,673 DEGs (Table S1-S10). Figure 3 represents the DEG distribution achieved using volcano plots (Figure 3A-3E). These plots effectively highlighted the upregulated (red), downregulated (blue), and non-significant (black) genes across the conditions, demonstrating the broad spectrum of transcriptional changes in the dataset. The results of these analyses provided the foundational data for identifying patterns of gene regulation and overlapping signatures between COPD and tuberculosis.
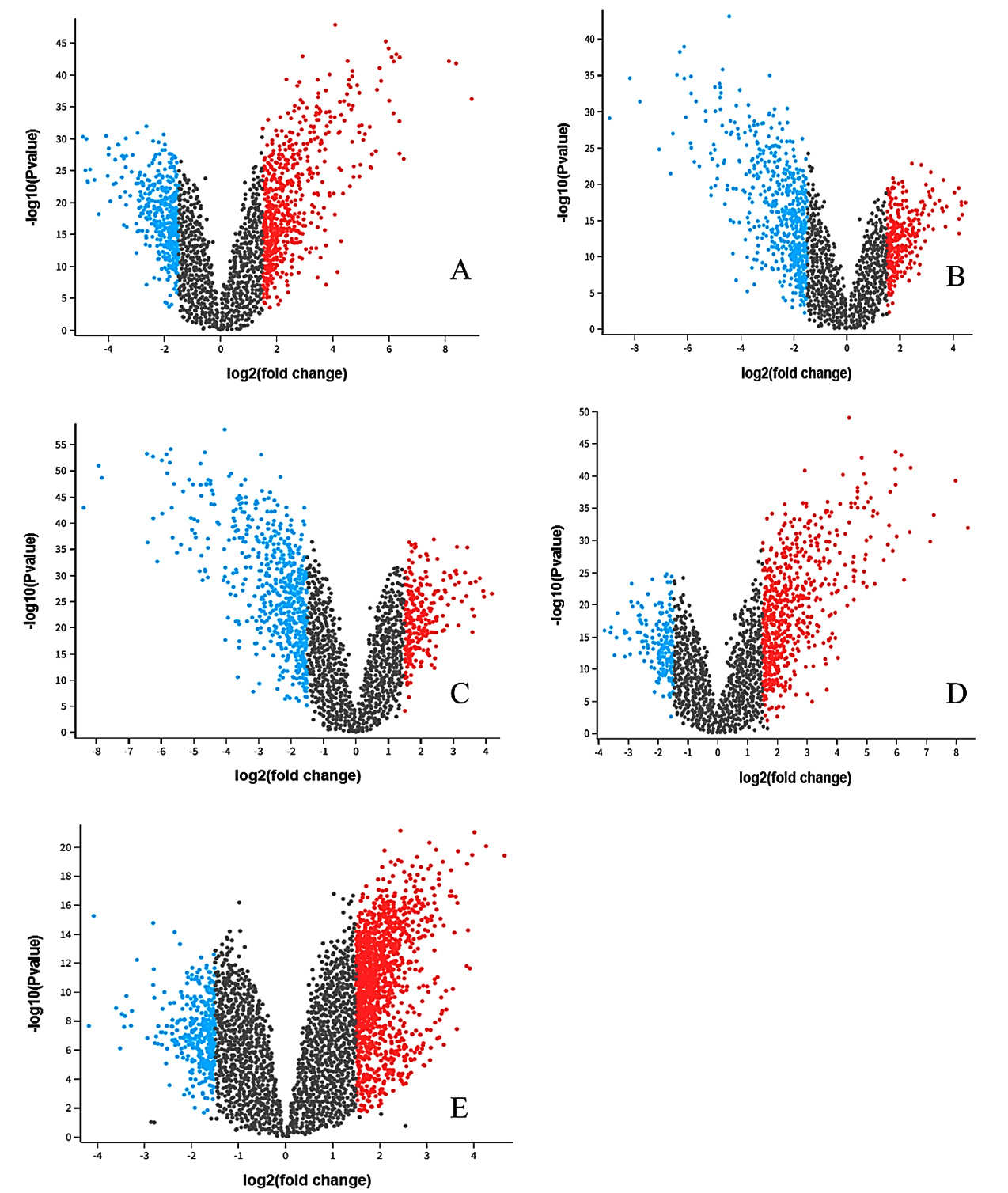
Figure 3. Volcano plots displaying differentially expressed genes across five comparison groups in the COPD and TB co-infection datasets: (A) COPD Unstimulated vs. COPD Stimulated, (B) COPD Stimulated vs. Smoker Unstimulated, (C) Smoker Stimulated vs. COPD Unstimulated, (D) Smoker Unstimulated vs. Smoker Stimulated, and (E) Healthy Control vs. Tuberculosis
A Venn diagram was generated to assess overlapping DEGs among the four COPD-related groups, revealing 670 shared DEGs (Table S11). These overlapping genes provided a crucial link for identifying common pathways and signatures in COPD. Further analysis of these shared DEGs in conjunction with the tuberculosis dataset uncovered 38 common genes between COPD and tuberculosis (Figure 4 and Table S9). These genes represent a shared molecular footprint between the two diseases, warranting deeper exploration to uncover their potential biological roles. This dual-level analysis, identifying DEGs within COPD conditions and then intersecting them with tuberculosis data, laid the foundation for downstream analyses, including functional annotation and pathway enrichment, to contextualize these genes in their respective biological processes.
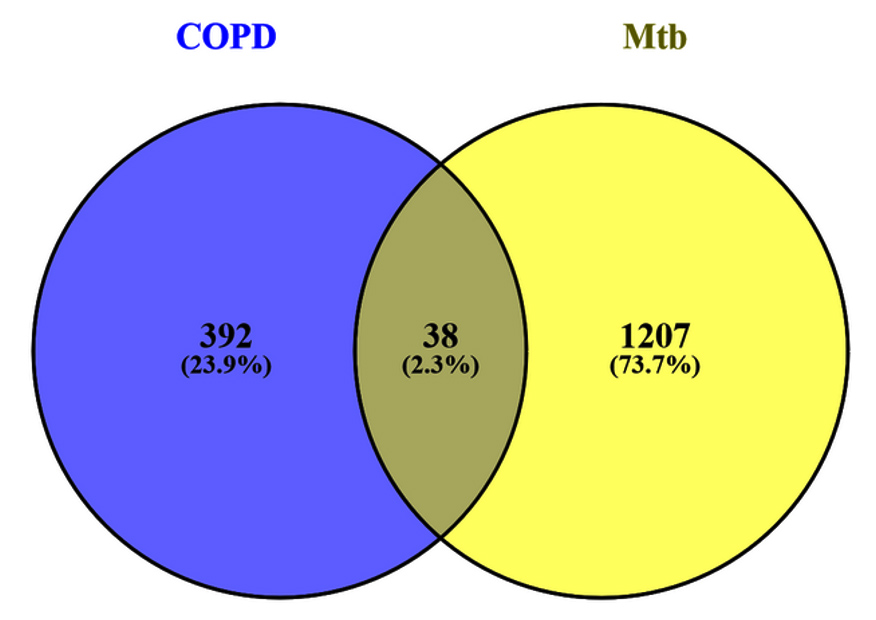
Figure 4. The image displays a Venn diagram. The right-hand side illustrates the common genes between Mtb and COPD
Commonly dysregulated genes in COPD and Mtb
Identifying 38 common genes between COPD and tuberculosis offers a valuable foundation for understanding the shared transcriptional signatures of these two conditions. These genes, identified through differential expression and comparative analysis, include notable candidates such as RELA, TNFAIP2, PLK3, RELB, and STAT1. Their involvement is consistent with active participation in different regulatory networks and pathways identified across the two diseases. For instance, genes such as RELA and RELB are essential to the NF-κB pathway, which controls inflammation signals and immune response activity. In contrast, TNFAIP2 and PLK3 contribute to stress reactions of the cell as well as mechanisms that control cell death; hence, their upregulation in both COPD and tuberculosis datasets is of interest. On the other hand, MS4A6A and IKBIP were found to be uniformly downregulated in all the conditions, suggesting that there are some common pathways active in modulating the immune system.
Adding to this foundation, a detailed comparison revealed nine genes consistently dysregulated across COPD, smoking, and tuberculosis datasets, reinforcing the hypothesis of shared molecular mechanisms. Seven of these genes specifically, RELA, RELB, PLK3, TNFAIP6, HIVEP3, EHD1, and PPARD demonstrated consistent upregulation in the configurations Smoker Stimulated vs COPD Unstimulated and COPD Stimulated vs Smoker Unstimulated (Figure 5). These genes manifest their functions in inflammation, cellular stress, and immune modulation. RELA and RELB, as central components of the NF-κB pathway, are pivotal in driving inflammation and immune responses,33 while PPARD and HIVEP3 highlight the interplay between metabolic regulation and immune signaling. PLK3 and TNFAIP6, meanwhile, underscore the chronic activation of cellular stress pathways in inflammatory states34 like COPD and tuberculosis.
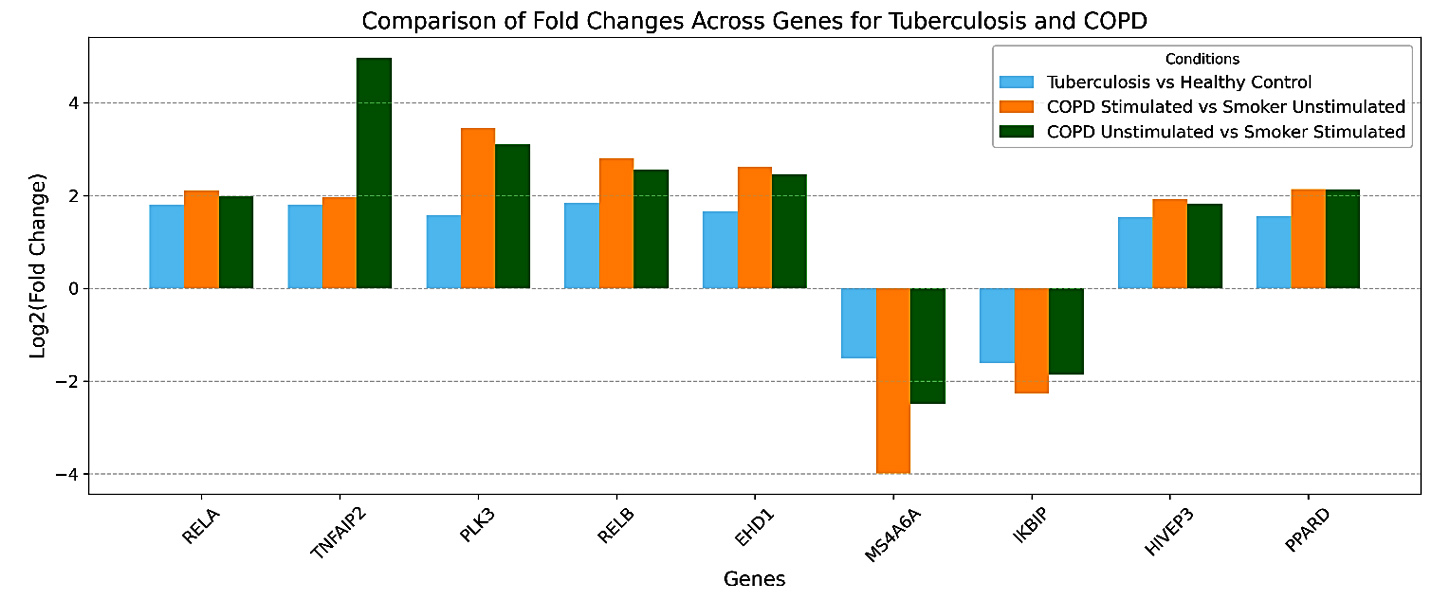
Figure 5. Top Dysregulated Genes Common to COPD and Mtb. Bar plots show fold changes for selected genes across three conditions: tuberculosis vs. healthy control (blue), COPD stimulated vs. smoker unstimulated (orange), and COPD unstimulated vs. smoker stimulated (green)
In contrast, IKBIP and MS4A6A were consistently downregulated across three comparisons (Figure 5), including COPD Unstimulated vs. COPD Stimulated and Smoker Unstimulated vs. Smoker Stimulated. IKBIP, a known regulator of NF-κB signaling, and MS4A6A, associated with immune cell activation, indicate impaired immune regulation across these conditions. Their downregulation aligns with chronic immune suppression observed in COPD and tuberculosis, further highlighting the convergence of dysregulated pathways in these diseases. The fold-change analysis supports these observations, as depicted in Figure 5. This analysis confirms the consistent trends in the upregulation of genes such as RELA, PLK3, PPARD, and HIVEP3, along with the suppression of IKBIP and MS4A6A across all three conditions. These patterns highlight and support common molecular mechanisms of chronic inflammatory diseases and the proposed functional significance of these genes in regulating immune responses, cellular stress, and metabolism.
Expanding upon this shared molecular basis, the comparative analysis of gene dysregulation between stimulated and unstimulated conditions in COPD and smokers reveals significant alignment patterns. When comparing Smoker Stimulated vs. COPD Unstimulated and COPD Stimulated vs. Smoker Unstimulated, the dysregulation trends appear remarkably consistent, with several genes demonstrating shared upregulation or downregulation patterns. For instance, RELA, PLK3, RELB, and TNFAIP2, known to play essential roles in inflammatory pathways, were upregulated under these conditions. This points to the chronic state of immune pathways activation using NF-κB signaling pathways in COPD conditions and conditions related to smokers. RELA and RELB are vital players in the NF-κB complex, regulating pro-inflammatory cytokine production, a hallmark of inflammation seen in both COPD and smoker-induced damage.
Moreover, TNFAIP2 and TNFAIP6 have a role in cellular stress responses and chronic inflammation, which underscores their participation in the molecular mechanisms of these diseases.35 Therefore, the comparison of the patterns of gene dysregulation in the context of these diseases reveals some important similarities and differences in the molecular mechanism of COPD and smokers’ immune system reactivity. The patterns identified in stimulated compared to unstimulated conditions highlight the role of pathways regulating activation and suppression: NF-κB signaling, interferon pathways, and metabolic processes appear as the core of the pathogenesis of these diseases. These findings lay the groundwork for exploring targeted interventions to modulate shared immune pathways in chronic inflammatory conditions.
Gene Ontology (GO) and pathway enrichment analysis
The functional roles of the 36 genes that are common to both COPD and Mtb infection have been investigated using Gene Ontology and pathway enrichment analysis approaches, showing that they have common functional roles. These analyses investigated three main GO categories: Biological Process (BP), Molecular Function (MF), and Cellular Component (CC) (Table S12-S14). The results highlight key pathways and mechanisms that may be important in these diseases’ immune response, inflammatory response, and cell metabolism. In the Biological Process (BP) category, the genes were significantly enriched in pathways related to the apoptotic process, cell differentiation, glucose metabolic processes, and responses to hypoxia (Figure 6). PPARD was highly present in more than one metabolic-related pathway, while, on the other hand, STAT1, FAS, and PLK3 were associated with apoptotic pathways. Apoptosis is an essential mechanism for maintaining immune system balance, and its disturbance may be involved in the inflammatory aspects of COPD and Mtb. Among the cellular mechanisms responding to hypoxia, RELA and PPARD genes are likely to have arisen as important tracks, presumably in reflection of the hypoxic microenvironments characteristic of diseased lung tissues.36 Considering that the experimental conditions frequently include anti-CD3 and anti-CD28 stimulation, such situations are explanatory as they represent T-cell receptor (TCR)-mediated activation and demonstrate how these genes may respond when there is immune stimulation or not.
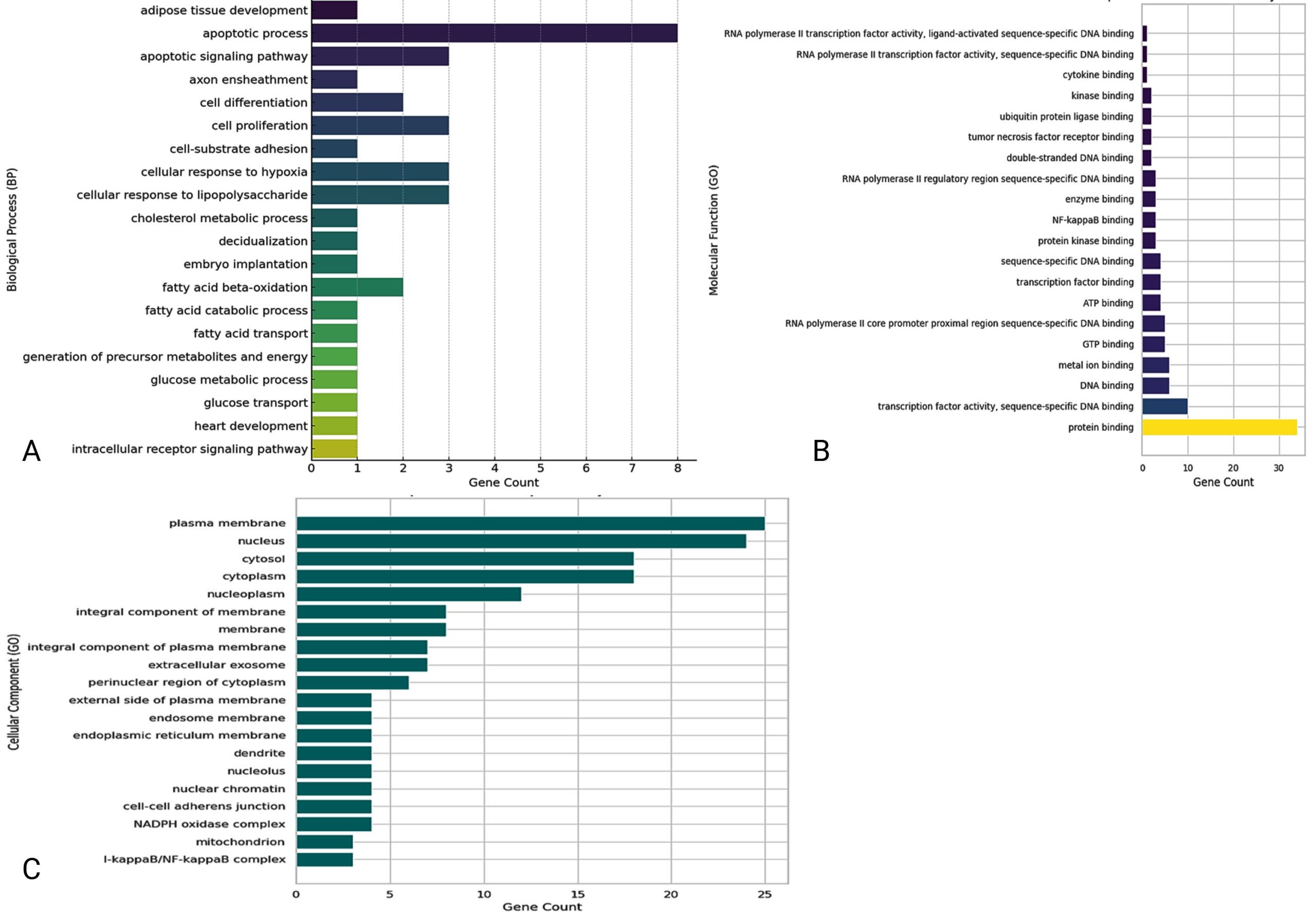
Figure 6. Gene Ontology and pathway enrichment analysis of genes associated with COPD and M. tuberculosis co-infection, providing a foundation for downstream analyses including functional annotation and biological interpretation. Panel (A) shows enrichment of genes in biological processes, panel (B) displays molecular function annotations, and panel (C) presents cellular component classifications
Molecular Function (MF) analysis revealed enrichment in transcription factor activity, DNA-dependent protein kinase activity, and protein-kinase activity binding (Figure 6). Among these genes, RELA, RELB, and STAT1 were crucial, which affirmed their importance in controlling gene transcription and signal transduction pathways. This pathway is known to affect the immune response and inflammation settings, key processes in COPD and tuberculosis. The involvement of STAT1 in cytokine signaling was also notable, as it could represent the molecular intersection where inflammatory signaling is amplified during anti-CD3 or anti-CD28 stimulation.37 In the Cellular Component (CC), enriched annotations were linked with the plasma membrane, nucleus, and cytosol (Figure 6). Genes such as GBP1, CYBB and CLEC4D were localized on the plasma membrane, a central component in immune receptor signaling. The nucleus, represented by genes like STAT1, RELA, and NBN, highlighted their roles in transcriptional regulation during immune activation. Interestingly, the NADPH oxidase complex, involving CYBB, was enriched, suggesting potential involvement in reactive oxygen species (ROS) generation, a process often implicated in both immune responses and tissue damage in COPD and Mtb. The pathway enrichment analysis indicated that these genes are also related to metabolic and inflammatory signaling pathways. For example, fatty acid metabolism and glucose transport pathways, mediated by PPARD, may indicate metabolic changes that occur in immune cells during chronic inflammation or infection.38 In addition, cytokine signaling pathways involving FAS, RELB, and STAT1 signalled their relevance to immune activation and apoptosis.
Functional insights through gene set and pathway enrichment analysis
The comprehensive enrichment analysis performed utilizing the Enrichr platform revealed the functional significance of the selected 38 genes in various biological contexts such as transcriptional factors, immune, cellular and disease ontology (Figure 7 and Table S15-S29). The analysis emphasizes their major involvement in immune, apoptosis and signaling processes, especially in inflammatory and infectious diseases like COPD and tuberculosis.
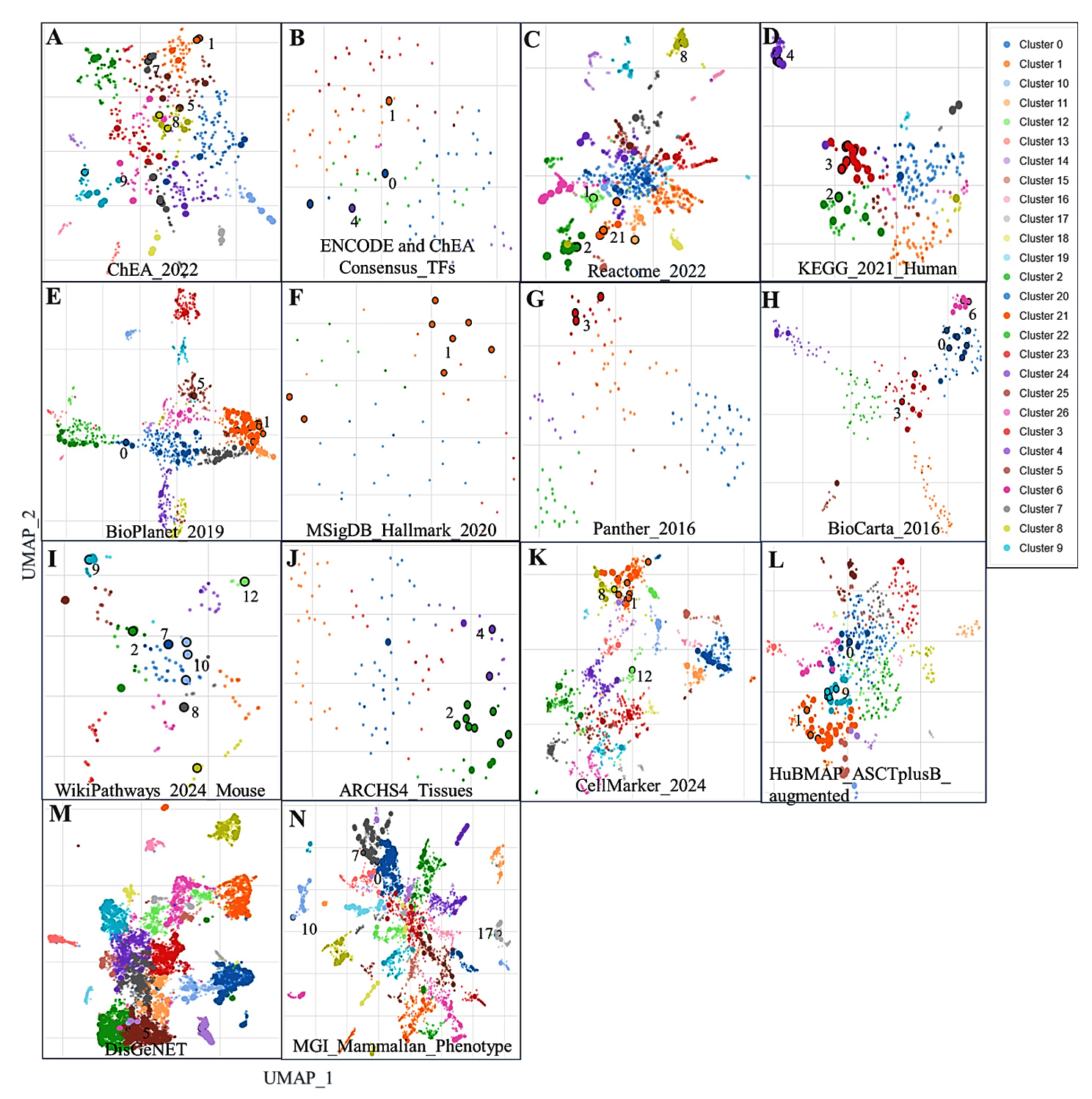
Figure 7. Scatterplot of gene set terms visualized using UMAP on TF-IDF values, with clusters identified by the Leiden algorithm. Point size and darkness indicate enrichment significance, and hovering reveals term details, p-values, and clusters. Panels: A-B (transcription factor-related DEGs), C-I (pathways), J-L (cell types/markers), M (disease-associated genes), N (gene ontology pathways)
The enrichment analysis results from ChEA 2022 and ENCODE libraries indicated significant enrichment for transcription factors like RELA, IRF8, and STAT5A. These are critical for the modulation of inflammation and immune-related signalling. The RELA gene were suppressed by core components of NF-κB signals such as those associated with STAT1, CD274, and FAS, which indicate essential functions that activate immune cells and promote inflammation (Figure 7). This directly relates to conditions like tuberculosis and COPD, where the immune system is dysregulated. Furthermore, the enriched transcription factor associations under anti-CD3/anti-CD28 stimulation conditions add strength to their association with T-cell activation and alteration of the immune response. Pathway enrichment analysis using Reactome 2022 and KEGG 2021 Human databases pinpointed key signaling pathways, including the C-type lectin receptor signaling pathway, NLRP3 inflammasome, and interferon-gamma signaling pathway. The genes CLEC4D, CLEC4E, and STAT1 are associated with innate immunity as they are enriched in the C-type lectin receptor signaling pathway. In the process, the involvement of genes such as GBP5, PSTPIP1, and CYBB also points out their other function of initiating inflammatory response through the NLRP3 inflammasome pathway. These pathways are particularly interesting in tuberculosis pathophysiology, considering the dominant role of innate immunity in curbing bacterial growth. Likewise, the contribution of STAT1, GBP5, and CYBB genes in adaptive immunity is also well depicted in the interferon-gamma signaling pathway.
The functional annotation of cellular markers through ARCHS4 and CellMarker datasets highlighted significant enrichment in immune cell types such as granulocytes, macrophages, and neutrophils. Other prominent clusters from these datasets were CYBB, CD274, GFAP, YWHAG, and GBP5, which can explain chronic inflammatory responses in tuberculosis and COPD. The functions of these genes correspond increased tuberculosis susceptibility, which has been associated with the functions of these genes at the level of macrophage-mediated pathogen clearance and inflammation in the pulmonary tissues. Disease-specific insights from DisGeNET and MGI Mammalian Phenotype annotations uncovered strong associations, especially with tuberculosis, and other autoimmune diseases. Genes such as STAT1, CD274, and CYBB emerged as key players linked to these conditions, emphasizing their roles in immune regulation and inflammation. Phenotypic annotations, including “abnormal macrophage physiology” and “lung inflammation,” further align with the involvement of genes like STAT1, FAS, and RELA in driving inflammatory responses in pulmonary diseases. The MSigDB Hallmark dataset enriched processes like interferon-gamma response, TNF-alpha signaling via NF-κB, and apoptosis, which are central to tuberculosis and COPD’s immune and inflammatory landscape (Figure 7). Genes such as TNFAIP6, PLK3, and FAS were key contributors to these pathways, indicating their involvement in apoptosis and immune regulation. In addition, BioPlanet datasets also suggested apoptosis-related processes, primarily through genes BCL2A1, FAS, and RELA, which associated cell death processes with apoptosis and other processes related to T cell immunity. Thus, integrating transcription factor binding, pathway enrichment, and cellular marker annotations highlights the complex roles of these 38 genes in immune signaling, inflammation, and apoptosis. Their enrichment across multiple datasets reflects their functional versatility and potential involvement in both innate and adaptive immune responses. While these results suggest broad regulatory roles, further experimental validation is necessary to delineate their exact contributions in the context of tuberculosis and COPD. These insights provide a foundation for understanding the shared molecular mechanisms underlying inflammatory and infectious diseases.
KEGG pathway analysis: Tuberculosis-specific insights
The KEGG pathway representation for tuberculosis (ko05152) emphasizes critical immune and molecular processes, with several highlighted genes shown in red, including STAT1 (hsa04630), Mincle (map05152), CNK (map04010), and NF-κB (hsa04064) (Figure 8). These genes are central to the host’s defense mechanisms and reflect key nodes in the immune signaling network against Mycobacterium tuberculosis. Their involvement builds upon the gene ontology and pathway enrichment findings, where immune processes and signaling pathways were recurrent themes across multiple analyses.
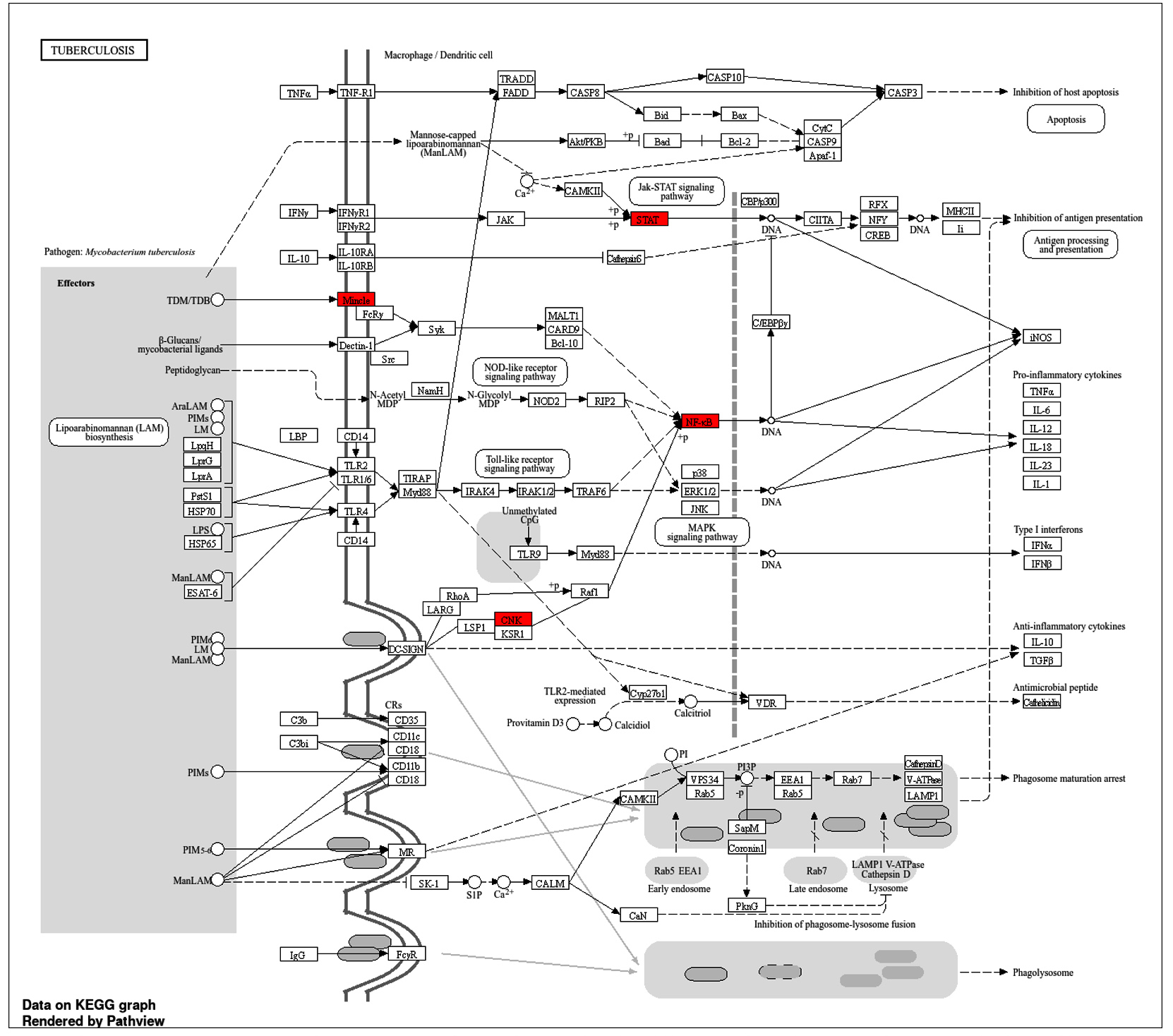
Figure 8. KEGG pathway map for tuberculosis, illustrating interactions and signaling cascades in macrophages and dendritic cells during infection with Mycobacterium tuberculosis. Nodes represent genes, with red-highlighted elements indicating key genes involved in the pathway
STAT1, a key component of the JAK-STAT signaling pathway, serves as a critical mediator of interferon-gamma (IFN-γ) signaling. The activation of STAT1 enhances the expression of genes responsible for antigen presentation and pro-inflammatory cytokines, linking this pathway to the broader immune network that combats M. tuberculosis. Its pivotal role in immune activation aligns with previous observations of its association with cytokine-mediated responses and apoptosis regulation. Mincle (CLEC4E), identified in the C-type lectin receptor signaling pathway, recognizes specific glycolipids from M. tuberculosis, such as trehalose dimycolate. The interaction of Mincle with mycobacterial components triggers downstream signaling cascades involving SYK and CARD9, leading to pro-inflammatory cytokine production, including IL-6 and TNF-α. This reinforces its role as a key immune sensor, bridging pathogen recognition with inflammatory responses. CNK is an important component that integrates the signals received from toll-like receptors (TLRs) with the MAPK pathway. Furthermore, this activation leads to the effective transmission of survival and immune signals, which leads to the production of cytokines. The involvement of CNK in modulating the MAPK pathway indicates a regulatory role of CNK-mediated signaling in maintaining immune homeostasis during infections.39 NF-κB as suggested, would be one of the more critical transcription factors in the immune response. Activated via the TLR and NOD-like receptor pathways, NF-κB engages in the transcription of pro-inflammatory cytokines and the genes necessary for apoptosis and cell survival (Figure 8). The pathway confirms NF-κB’s crucial role in the enhancement of immune response against Mycobacterium tuberculosis while at the same time observing its plausible role in the evasion strategies of the pathogen.40 The integration of these signaling components shows the complexity of host response to M. tuberculosis infection, whereby the engagement of these pathways, particularly STAT1, Mincle, CNK, and NF-κB, contributes significantly towards regulating immune responses. These results fit well with the overall picture of enriched immune pathways described above, including interferons, antigen presentation, and cytokine-triggered responses.41 The convergence of these pathways underlines their interconnected roles in orchestrating the immune response and balancing pathogen control with host tissue preservation.
Protein-protein interaction networks and key molecular complexes
The protein-protein interaction (PPI) analysis offers a detailed view of the molecular landscape connecting genes associated with COPD and Mycobacterium tuberculosis (Mtb). The network comprises 37 nodes and 55 edges, revealing crucial molecular interactions and regulatory hubs. A significant finding of this analysis is the identification of an MCODE module z, which highlights a dense cluster of interconnected genes, namely GBP5, GBP1, STAT1, IFIH1, GBP3, CD274, IDO1, and LAP3 (Figure 9). These genes form the central regulatory unit, indicating their pivotal roles in immune modulation and inflammatory responses, which are critical in both COPD and Mtb pathophysiology. The presence of STAT1, a well-established transcription factor regulating interferon and cytokine responses, reinforces its central role in mediating immune responses.
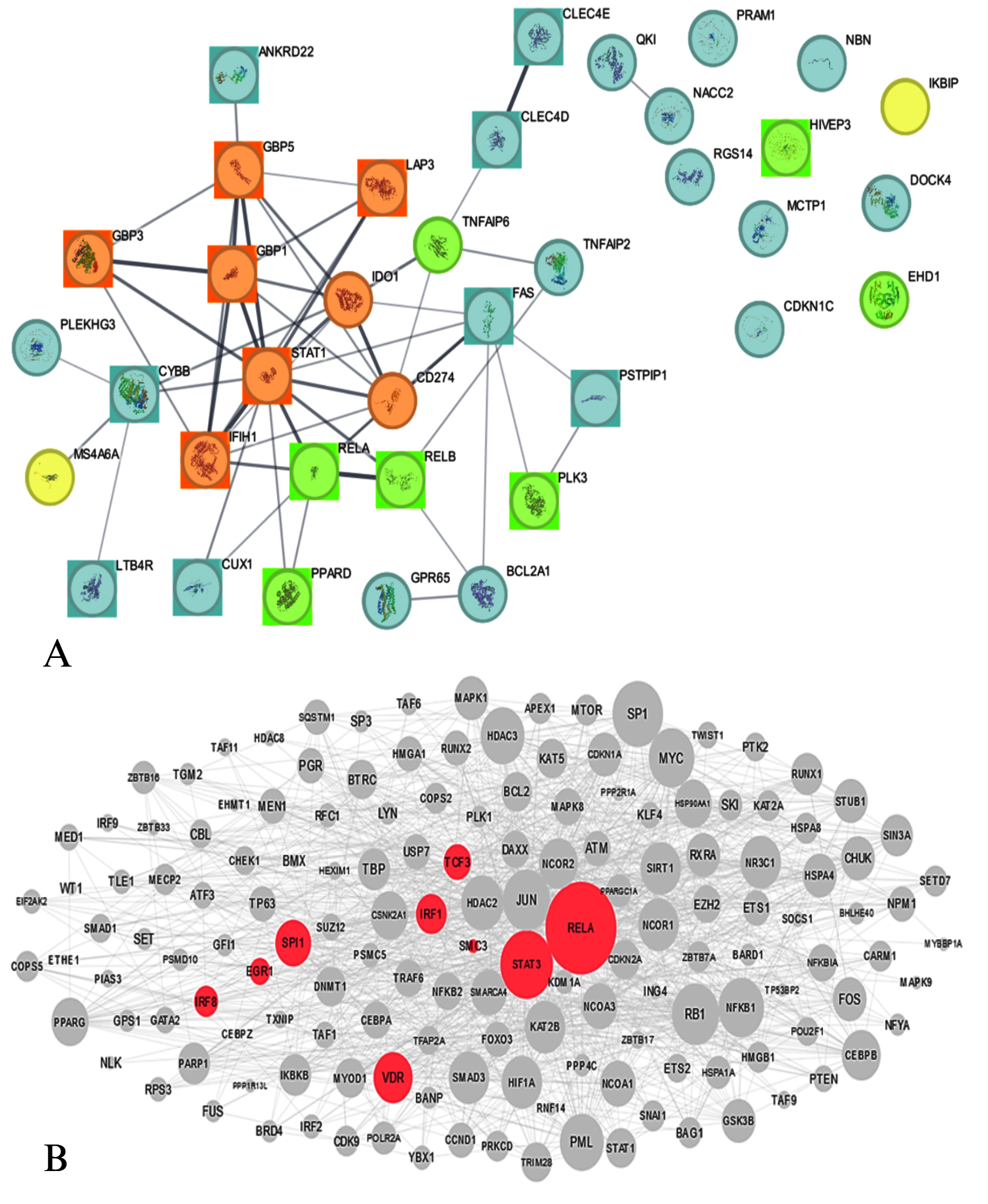
Figure 9. Protein–protein interaction (PPI) network analysis of pathways and associated transcriptional regulation. Panel (A) displays the PPI network, with nodes and edges color-coded according to functional annotations: green nodes represent up-regulated genes, yellow nodes indicate down-regulated genes, square nodes highlight experimentally validated genes, and brown nodes denote high-scoring MCODE cluster genes. Panel (B) shows upstream transcription factors associated with the dysregulated genes, as identified through regulatory network analysis
As observed in the network, the upregulation of genes such as PPARD, RELA, RELB, PLK3, TNFAIP6, HIVEP3, and EHD1 suggests an active involvement in pathways linked to inflammation, immune signaling, and tissue remodeling. For instance, RELA and RELB, core components of the NF-κB complex, are known to regulate pro-inflammatory cytokines, aligning with earlier observations of these pathways in the KEGG and GO analyses. Their interaction with STAT1 further supports the hypothesis that shared regulatory circuits drive inflammatory and immune processes in COPD and Mtb. The co-expression and interaction evidence between RELA and STAT1 (combined score: 0.864) and GBP1 and GBP5 (combined score: 0.908) emphasize their collaborative roles in modulating host defense mechanisms. These connections echo the findings from previous sections where immune-related pathways, such as interferon signaling and NF-κB activation, were enriched.
Genes IKBIP and MS4A6A, which seem to be associated with immune evasion strategies of Mtb, are found to be downregulated in the shared network and, therefore, might be considered suppressed pathways. The decreased expression of these genes can indicate that the pathogen alters its behavior in such a way as to allow it to remain within the host, which would explain the presence of the gene. Interestingly, this finding reinforces the KEGG analysis of tuberculosis pathways in which the genes STAT1 and NF-κB were marked among the important immunological response genes toward Mtb infection. Some empirical data from STRING (Tables S30-S31), supported with connections GbP1-GbP3 (score: 0.936), and GbP1-IFIH1 (score: 0.87), offer further corroboration of the role that guanylate-binding proteins (GBPs) play as central figures in the protective responses of hosts. These proteins are known to play roles in pathogen detection and immune signaling, further reinforcing their functional importance in the shared COPD-Mtb network.
Similarly, the interaction between RELA and RELB (combined score: 0.999) points to the activation of NF-κB-mediated pathways, which were previously highlighted as key mechanisms in both diseases. To further elucidate the regulatory hierarchy, the X2K Appyter tool was employed to predict upstream transcription factors associated with the dysregulated genes (Table S32). This analysis identified RELB, IRF1, IRF7, and NFKB2 as significant regulators. RELB and NFKB2, as components of the NF-κB pathway, align with their roles as central hubs in the PPI network (Figure 9). Notably, IRF1 and IRF7, transcription factors involved in interferon signaling, were linked to STAT1, CD274, and GBP family genes, which were consistently downregulated across conditions. This highlights a suppression of interferon-regulated pathways in COPD and tuberculosis.
Tissue-specific and single-cell gene expression analysis
The exploration of tissue-specific expression patterns for dysregulated genes provides critical insights into their functional roles in COPD and tuberculosis, with lung and blood tissues emerging as primary sites of transcriptional activity (Figure 10). The heatmap analysis highlights genes such as RELA, TNFAIP2, PLK3, RELB, and TNFAIP6, which exhibited markedly higher expression in lung tissues. These findings emphasize their involvement in immune and inflammatory processes within the respiratory system. For example, RELA and RELB, key components of the NF-κB pathway, had median TPM values of 101.8 and 31.52, respectively, underscoring their roles in modulating pro-inflammatory cytokines. Conversely, IKBIP and MS4A6A, which were consistently downregulated, showed median TPM values of 11.20 and 41.33, respectively, suggesting their involvement in immune suppression and impaired signaling pathways in chronic inflammation. Similarly, metabolic regulator PPARD and stress response gene PLK3 demonstrated prominent expression in lung tissues, with median TPM values of 46.77 and 39.97, highlighting their roles in metabolic and stress adaptation within the lung microenvironment.
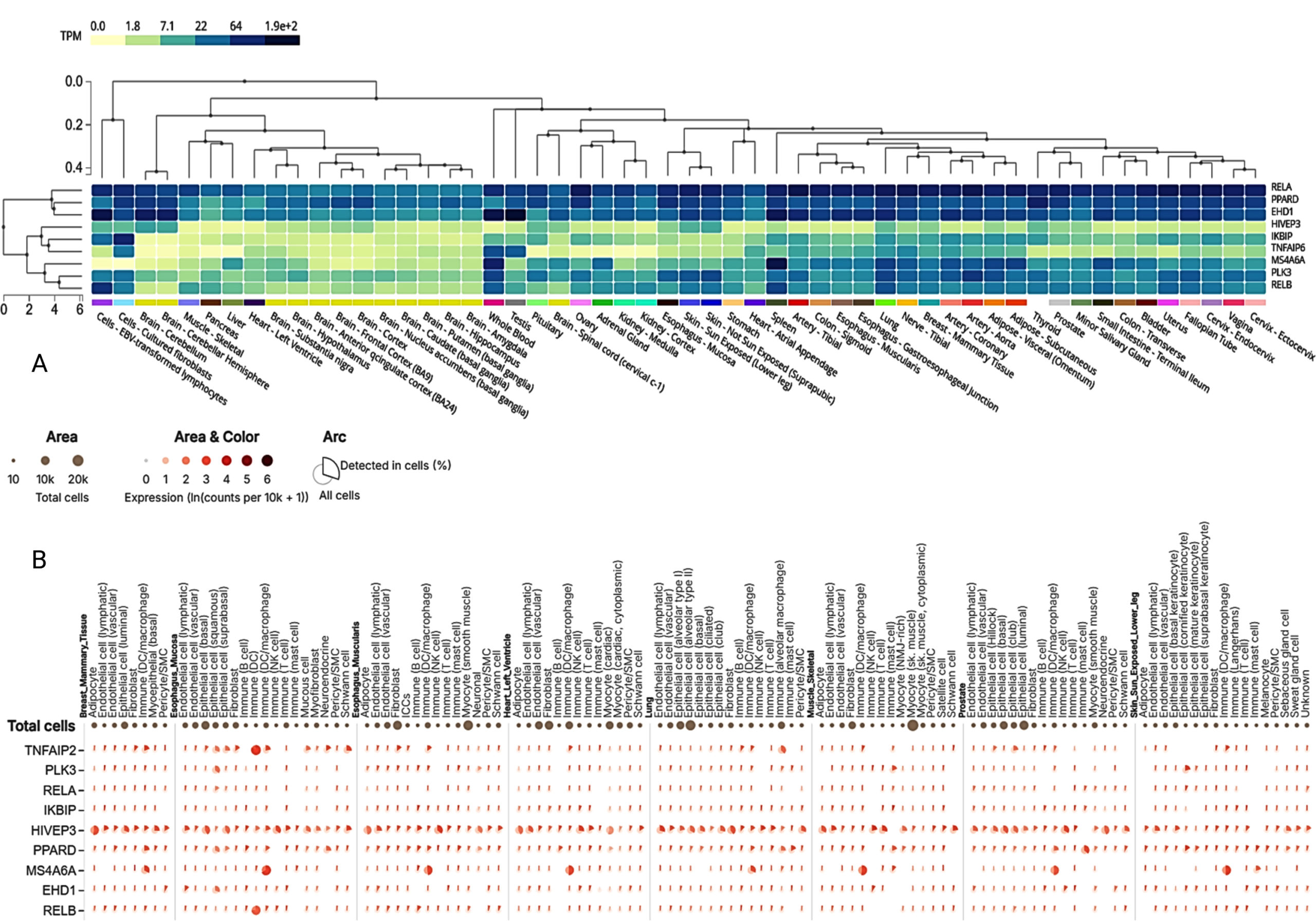
Figure 10. Cluster analysis of tissue-specific expression patterns of a common gene across co-infections is shown in panel (A), highlighting variations in gene expression across different tissues. Panel (B) presents single-cell RNA sequencing analysis of lung tissue, providing detailed insights into gene expression at the single-cell level
On the other hand, some genes showed greater expression in whole blood expression profiles. Some systemic immune genes, including EHD1, MS4A6A, and TNFAIP2, had higher median TPM values in whole blood. In this regard, it is interesting to note that EHD1 was reported to have the highest median TPM value of 126.6. This suggests that EHD1 is associated with mechanisms such as leukocyte trafficking, which are important for triggering systemic inflammation. Similarly, TNFAIP2, with a median TPM value of 81.22, highlights its dual role in localized pulmonary inflammation and systemic immune modulation. Tissue-specific divergence was observed for specific genes, such as RELA and RELB, which were prominently expressed in lung tissues but moderately expressed in whole blood (mean TPM values of 59.59 and 26.53, respectively) (Figure 10). Such divergence demonstrates the existence of tissue-specific regulatory functions of these genes about adaptation to a pulmonary microenvironment and systemic immunity.
The PD-L1 (CD274) expression in lung and blood samples further exemplifies the interaction between immune checkpoint control and inflammation. This observation aligns with prior findings of immune exhaustion and evasion mechanisms in COPD and tuberculosis, reinforcing the importance of immune checkpoint pathways in regulating inflammation. The expression patterns of RELA, RELB, TNFAIP2, TNFAIP6, and PLK3 across tissues justify their importance in pulmonary diseases, whereas their circulation in blood indicates their wider disease involvement. These discussions help provide a scientific rationale for formulating strategies that can simultaneously address the abnormalities at the tissue and the system levels, as seen in the cases of COPD and tuberculosis. In addition, a detailed investigation of scRNA-seq data analysis demonstrated that many of the dysregulated genes in the lung tissue were expressed in an ectopic manner in a cell type-specific manner (Figure 10). For instance, HIVEP3, PPARD, TNFAIP2, MS4A6A, RELA, and RELB were listed among the genes with the highest expression in specific cell types, thus pinpointing the involvement in the processes that lead to COPD. HIVEP3 was predominantly found in alveolar type II epithelial cells and macrophages with a mean expression of 2.148 and 2.536, respectively, to suggest that it maintains epithelial integrity and immune response. Likewise, PPARD revealed substantial presence in alveolar macrophages and mast cells with mean expression of 1.935 and 2.654, respectively, implying their role in immune and metabolic regulation. MS4A6A, another key gene, exhibited robust expression in macrophages (mean expression 2.625), providing further evidence regarding its function in innate immunity, whereas TNFAIP2 appeared to be expressed predominantly in alveolar macrophages (mean expression 2.201), suggestive of its pro-inflammatory effects.
Combining tissue-specific and single-cell data completes the picture of dysregulation genes in COPD. For instance, during this analysis, genes like RELA, TNFAIP2, and RELB were shown to be overexpressed in both lung tissue and the respective cell types, implying that they play important roles in regulating immune and inflammatory responses. On the contrary, IKBIP and MS4A6A, known to be repressed in the preceding studies, showed low expression in certain immune cells, indicating that they are part of the inactive immune pathways. The overexpression of the immune checkpoint marker CD274 (PD-L1) in immune cells suggests that there are mechanisms to get rid of immune over-activation, one of the characteristics of diseases with chronic inflammation, such as COPD. The expression of the immune receptor phenotype genes is not uniform, suggesting functional differences among these genes. For instance, HIVEP3 and TNFAIP2, prominent in alveolar macrophages, have roles in tissue homeostasis and modulating the local immune response in chronic inflammation. Similarly, the expression of PPARD in mast cells and macrophages highlights its dual role in regulating metabolic and immune processes, making it a potential therapeutic target.42 By integrating findings from tissue-specific and single-cell analyses, this study provides a detailed framework for understanding the transcriptional dynamics in COPD and tuberculosis, paving the way for future therapeutic strategies targeting tissue- and cell-specific pathways.
Summary and limitation of study
This study aimed to explore the shared and distinct molecular mechanisms underlying COPD and tuberculosis, focusing on transcriptional dysregulation, pathway enrichment, protein-protein interactions, and tissue-specific and single-cell gene expression. Using a systematic and integrated approach to the analysis of expressed genes (DEG) for stimulated and unstimulated conditions, 38 common genes were identified as central to these devastating diseases’ pathophysiology. These findings give insight into the molecular cores that intertwine chronic inflammatory and immune processes in COPD and tuberculosis.
Key genes such as RELA, RELB, PLK3, TNFAIP2, and STAT1 have become pivotal in immune signaling, inflammatory response, and cellular stress responses. The expression of these genes under all analyzed conditions indicates their involvement in chronic inflammation and immune dysregulation. On the other hand, genes such as IKBIP and MS4A6A were expressed at lower levels, which may indicate that these genes can be present in downregulated immune pathways that lead to immune evasion. Pathway analysis revealed that other critical processes modulating the cells include those mediated by NF-κB and other cytokines and metabolic alteration, which is some of the mechanisms through which the immune system is activated. Inflammation takes place in the affected tissues in these diseases. It was also possible to see a more generalized picture of the molecular landscape from PPI networks that include major gene hubs such as STAT1, RELA, GBP5, and GBP1 as essential regulation elements. Tighter and more dependent filamentous arrangements of these genes draw greater attention to their potential roles in the control mechanisms of immune responsiveness. Clustering transcription factors such as RELB, IRF1, and NFKB2 as transcription factors converging on the PIC revealed an upstream regulation of these pathways in a sequence. In particular, these findings provide further proof supporting the involvement of immune signaling, apoptosis, metabolism, and other biological processes in the common molecular pathogeny of COPD and tuberculosis.
Tissue-specific expression analysis identified the lungs and whole blood as primary sites of transcriptional activity for the dysregulated genes. Notably, genes such as RELA, PLK3, and TNFAIP2 were prominently expressed in lung tissues, emphasizing their roles in pulmonary inflammation and immune responses. Complementary findings from whole blood analysis identified genes such as EHD1 and MS4A6A as significant contributors to systemic immune regulation that are likely involved in leukocyte trafficking and systemic inflammation. These tissue-specific patterns suggest that while certain genes are critical to localized pulmonary processes, other genes that are equally important but their roles are primarily systemic, are also present due to the heterogeneous nature of these diseases. Single-cell RNA sequencing (scRNA-seq) added depth to this analysis by revealing cell-type-specific expression patterns. For instance, HIVEP3, PPARD, and even TNFAIP2 have markedly higher expression in alveolar macrophages and epithelium – indicating the genes’ involvement in the local immune response and tissue balance. Similarly, the expression of CD274 (PD-L1) in immune cells was also consistent, indicating a possible role of immune checkpoint regulation in chronic inflammation.43 These findings highlight the need to understand transcriptional control at the levels of the tissue as well as every cell type in order to reveal pathogenetic mechanisms.
The purpose of this work was not only to identify shared molecular signatures but also to contextualize these findings within the broader framework of disease biology. The information provided allows one to target more focused approaches to designing major players involved in the inflammation cascade and immune dysregulation associated with COPD and tuberculosis comorbidity. However, this study is limited by its focus on bioinformatics, which highlights why experimental studies are needed to substantiate the functional relevance of the identified targets. The approaches were purely based on transcriptional changes, with no regard for post-transcriptional or post-translational modification, which are essential in regulating genes. In addition, the absence of experimental evidence also limits one’s ability to substantiate the functional roles of identified genes. In addition, the temporal aspects of these dysregulated pathways remain elusive and need more detailed investigation to discover how they develop and their clinical significance. Thus, this study extends the existing literature on transcriptional profiles between COPD and tuberculosis and the molecular pathways involved, explicitly focusing on NF kappa B signaling, immune checkpoints, and metabolic dysregulation of these diseases. These data provide a basis for developing targeted therapies to alter critical regulators of inflammation and immune responses. In future studies, longitudinal and multi-omics approaches should be applied to investigate the complexity of these diseases and their common pathophysiology to validate these pathways experimentally.
The co-infection of tuberculosis and COPD represents a significant problem due to their overlapping inflammatory and immune dysregulation features. This study aimed to elucidate common functional pathways, with a focus on identifying transcriptional signatures and their role in disease progression. In total, 38 shared genes were identified during the integrative analysis related to activating several pathways e.g., NF-κB signaling, immune checkpoint regulation, and metabolic pathways. Upregulated genes like RELA, RELB, and PLK3 emphasized persistent inflammation, while downregulated genes such as IKBIP and MS4A6A suggested impaired immune signaling. Functional and tissue-specific analyses reinforced the importance of these genes in lung pathology and systemic immune response, particularly in macrophages and alveolar epithelial cells. These results show the molecular relationship between COPD and tuberculosis and their cross relationship, depicting the pathological features shared by both diseases. Although this study provides a solid basis for the investigation of mechanisms of co-infection, further study is needed to test these concepts experimentally to develop treatment strategies. This work lays the foundation for future research targeting immune and inflammatory pathways in COPD and tuberculosis co-infection.
Additional file: Additional Table S1-S32.
ACKNOWLEDGMENTS
The authors extend their appreciation to Northern Border University, Saudi Arabia, for supporting this work through project number NBU-CRP-2025-2042.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
AUTHORS’ CONTRIBUTION
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
FUNDING
This study was supported by the Northern Border University, Saudi Arabia.
DATA AVAILABILITY
All datasets generated or analyzed during this study are included in the manuscript and its supplementary file.
ETHICS STATEMENT
Not applicable.
- Gai X, Allwood B, Sun Y. Advances in the awareness of tuberculosis-associated chronic obstructive pulmonary disease. Chinese Med J Pulm Crit Care Med. 2024;2(4):250-256.
Crossref - Sarkar M, Srinivasa, Madabhavi I, Kumar K. Tuberculosis associated chronic obstructive pulmonary disease. Clin Respir J. 2017;11(3):285-295.
Crossref - Lu W, Aarsand R, Schotte K, et al. Tobacco and COPD: presenting the World Health Organization (WHO) Tobacco Knowledge Summary. Respir Res. 2024;25(1):338.
Crossref - Imran M, Alshrari AS, Hafiz MN, et al. Exploring therapeutic paradigm focusing on genes, proteins, and pathways to combat leprosy and tuberculosis: A network medicine and drug repurposing approach. J Infect Public Health. 2025;18(6):102763.
Crossref - Rahlwes KC, Dias BRS, Campos PC, Alvarez-Arguedas S, Shiloh MU. Pathogenicity and virulence of Mycobacterium tuberculosis. Virulence. 2023;14(1):2150449.
Crossref - Chakrabarti B, Calverley PMA, Davies PDO. Tuberculosis and its incidence, special nature, and relationship with chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2007;2(3):263-272.
- O’Toole RF, Shukla SD, Walters EH. TB meets COPD: An emerging global co-morbidity in human lung disease. Tuberculosis. 2015;95(6):659-663.
Crossref - Zhuang L, Yang L, Li L, Ye Z, Gong W. Mycobacterium tuberculosis: immune response, biomarkers, and therapeutic intervention. MedComm. 2024;5(1):e419.
Crossref - Alfahad AJ, Alzaydi MM, Aldossary AM, et al. Current views in chronic obstructive pulmonary disease pathogenesis and management. Saudi Pharm J. 2021;29(12):1361-1373.
Crossref - Rodrigues S de O, da Cunha CMC, Soares GMV, Silva PL, Silva AR, Goncalves-De-albuquerque CF. Mechanisms, Pathophysiology and Currently Proposed Treatments of Chronic Obstructive Pulmonary Disease. Pharm. 2021;14(10):979.
Crossref - Carabali-Isajar ML, Rodriguez-Bejarano OH, Amado T, et al. Clinical manifestations and immune response to tuberculosis. World J Microbiol Biotechnol. 2023;39(8):206.
Crossref - Ehlers S, Schaible UE. The granuloma in tuberculosis: Dynamics of a host-pathogen collusion. Front Immunol. 2012;3:38036.
Crossref - Inghammar M, Ekbom A, Engstrom G, et al. COPD and the Risk of Tuberculosis – A Population-Based Cohort Study. PLoS One. 2010;5(4):e10138.
Crossref - Kayongo A, Nyiro B, Siddharthan T, et al. Mechanisms of lung damage in tuberculosis: implications for chronic obstructive pulmonary disease. Front Cell Infect Microbiol. 2023;13:1146571.
Crossref - Barnes PJ, Burney PGJ, Silverman EK, et al. Chronic obstructive pulmonary disease. Nat Rev Dis Prim. 2015;1:15076.
Crossref - Slobodyanyuk M, Bahcheli AT, Klein ZP, Bayati M, Strug LJ, Reimand J. Directional integration and pathway enrichment analysis for multi-omics data. Nat Commun. 2024;15:5690.
Crossref - Wang X, Fan D, Yang Y, Gimple RC, Zhou S. Integrative multi-omics approaches to explore immune cell functions: Challenges and opportunities. iScience. 2023;26(4):106359.
Crossref - Joshi A, Kumar A, Kaushik V. Functional Genomics and Network Biology. In: Singh, V., Kumar, A. (eds) Advances in Bioinformatics. Springer, Singapore. 2024:71-96.
Crossref - Helmy M, Smith D, Selvarajoo K. Systems biology approaches integrated with artificial intelligence for optimized metabolic engineering. Metab Eng Commun. 2020;11:e00149.
Crossref - Clough E, Barrett T. The Gene Expression Omnibus Database. In: Mathé, E., Davis, S. (eds) Statistical Genomics. Methods in Molecular Biology, vol 1418. Humana Press, New York, NY. 2016;1418:93.
Crossref - Maertzdorf J, Weiner J, Mollenkopf HJ, et al. Common patterns and disease-related signatures in tuberculosis and sarcoidosis. Proc Natl Acad Sci U S A. 2012;109(20):7853-7858.
Crossref - Zhu M, Ye M, Wang J, Ye L, Jin M. Construction of Potential miRNA-mRNA Regulatory Network in COPD Plasma by Bioinformatics Analysis. Int J Chron Obstruct Pulmon Dis. 2020;15:2135-2145.
Crossref - Foltz SM, Greene CS, Taroni JN. Cross-platform normalization enables machine learning model training on microarray and RNA-seq data simultaneously. Commun Biol. 2023;6(1):222.
Crossref - Zhao Y, Wong L, Goh WW Bin. How to do quantile normalization correctly for gene expression data analyses. Sci Rep. 2020;10:15534.
Crossref - Chen X, Sarkar SK. On Benjamini-Hochberg procedure applied to mid p-values. J Stat Plan Inference. 2020;205:34-45.
Crossref - Li T, Zou Y, Li X, Wong TKF, Rodrigo AG. Mugen-UMAP: UMAP visualization and clustering of mutated genes in single-cell DNA sequencing data. BMC Bioinformatics. 2024;25(1):308.
Crossref - Kanehisa M, Goto S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000;28(1):27-30.
Crossref - Croft D, O’Kelly G, Wu G, et al. Reactome: a database of reactions, pathways and biological processes. Nucleic Acids Res. 2010;39(Database issue):D691.
Crossref - Chen EY, Tan CM, Kou Y, et al. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013;14(1):128.
Crossref - Szklarczyk D, Franceschini A, Kuhn M, et al. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011;39:D561-D568.
Crossref - Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498-2504.
Crossref - Keen JC, Moore HM. The Genotype-Tissue Expression (GTEx) Project: Linking Clinical Data with Molecular Analysis to Advance Personalized Medicine. J Pers Med. 2015;5(1):22.
Crossref - Yu H, Lin L, Zhang Z, Zhang H, Hu H. Targeting NF-κB pathway for the therapy of diseases: mechanism and clinical study. Signal Transduct Target Ther. 2020;5(1):209.
Crossref - Chu E, Mychasiuk R, Hibbs ML, Semple BD. Dysregulated phosphoinositide 3-kinase signaling in microglia: shaping chronic neuroinflammation. J Neuroinflammation. 2021;18(1):276.
Crossref - Jin G, Liu Y, Xu W, et al. Tnfaip2 promotes atherogenesis by enhancing oxidative stress induced inflammation. Mol Immunol. 2022;151:41-51.
Crossref - Ortmann B, Druker J, Rocha S. Cell cycle progression in response to oxygen levels. Cell Mol Life Sci. 2014;71(18):3569-3582.
Crossref - Manaithiya A, Bhowmik R, Bhattacharya K, et al. A cheminformatics and network pharmacology approach to elucidate the mechanism of action of Mycobacterium tuberculosis g-carbonic anhydrase inhibitors. Front Pharmacol. 2024;15:1457012.
Crossref - Montaigne D, Butruille L, Staels B. PPAR control of metabolism and cardiovascular functions. Nat Rev Cardiol. 2021;18(12):809-823.
Crossref - Maisonneuve P, Sahmi M, Bergeron-Labrecque F, et al. The CNK-HYP scaffolding complex promotes RAF activation by enhancing KSR-MEK interaction. Nat Struct Mol Biol. 2024;31(7):1028-1038.
Crossref - Guo Q, Jin Y, Chen X, et al. NF-κB in biology and targeted therapy: new insights and translational implications. Sig Transduct Target Ther. 2024;9(1):53.
Crossref - Alam Z, Devalaraja S, Li M, et al. Counter Regulation of Spic by NF-κB and STAT Signaling Controls Inflammation and Iron Metabolism in Macrophages. Cell Rep. 2020;31(13):107825.
Crossref - Toobian D, Ghosh P, Katkar GD. Parsing the Role of PPARs in Macrophage Processes. Front Immunol. 2021;12.
Crossref - Suarez GV, Melucci Ganzarain C del C, Vecchione MB, et al. PD-1/PD-L1 Pathway Modulates Macrophage Susceptibility to Mycobacterium tuberculosis Specific CD8+T cell Induced Death. Sci Reports. 2019;9(1):1-14.
Crossref
© The Author(s) 2025. Open Access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License which permits unrestricted use, sharing, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.