


ISSN: 0973-7510
E-ISSN: 2581-690X
Halophilic archaeon Halolamina pelagica CDK2, showcasing plant growth-promoting properties and endurance towards harsh environmental conditions (high salinity, heavy metals, high temperature and UV radiation) was sequenced earlier. Pan-genome of Halolamina genus was created and investigated for strain-specific genes of CDK2, which might confer it with features helping it to withstand high abiotic stress. Pathways and subsystems in CDK2 were compared with other Halolamina strain CGHMS and analysed using KEGG and RAST. A genome-scale metabolic model was reconstructed from the genome of H. pelagica CDK2. Results implicated strain-specific genes like thermostable carboxypeptidase and DNA repair protein MutS which might protect the proteins and DNA from high temperature and UV denaturation respectively. A bifunctional trehalose synthase gene responsible for trehalose biosynthesis was also annotated specifying the need for low salt compatible solute strategy, the probable reason behind the ability of this haloarchaea to survive in a wide range of salt concentrations. A modified shikimate and mevalonate pathways were also identified in CDK2, along with many ABC transporters for metal uptakes like zinc and cobalt through pathway analysis. Probable employment of one multifunctional ABC transporter in place of two for similar metals (Nickel/cobalt and molybdenum/tungsten) might be employed as a strategy for energy conservation. The findings of the present study could be utilized for future research relating metabolic model for flux balance analysis and the genetic repertoire imparting resistance to harsh conditions can be transferred to crops for improving their tolerance to abiotic stresses.
Halolamina pelagica, Halophilic Archaea, Genome-scale metabolic model reconstruction, Osmotic stress adaptation
Until 1970, archaebacteria were placed in the domain “Bacteria”. It was only after 1970, that these microorganisms were grouped into the third domain of life called the “Archaea”.1 Amongst archaea, halophilic archaea have acquired considerable attention because of their ability to withstand high salinity. Not only extreme salinity but these halophilic archaea are also known to escape high metal toxicity, low levels of oxygen and high UV rays.2 Their existence has also been reported in the extra-terrestrial environment.3 The Rann of Kutch, Gujarat, India experiences extremely hot climate (50-55°C), severe UV radiation, and metal toxicity. This makes Rann of Kutch a suitable niche for analysing genes and pathways present in native extremophilic microorganisms which assist them in their adaptation and survival. Earlier we had reported the presence of P solubilizing and phytohormone producing halophilic archaeon, Halolamina pelagica CDK2 in the rhizosphere of wild vegetation inhabiting the hypersaline soils of Rann of Kutch, Gujarat, India.4,5 Based on its ability of tolerant to hyper salinity and plant growth promoting ability we carried out its whole genome sequencing (NCBI GenBank Accession No. LGUC00000000)6 with a view to understand its genetic mechanism of tolerance to different abiotic stresses, their subsequent mining and utilization. Previous studies carried out for the class Halobacteria on their core genome, pan genome and ancestral state reconstruction revealed high percentage of heterogeneity among halophilic archaea.7 Hence, there is a need to carry out a comprehensive analysis of genome, genes, and pathways of halophilic archaea Halolamina pelagica CDK2 with a special emphasis on its ability to tolerate different kinds of abiotic stresses. One way of studying these strategies is through the construction of a genome-scale metabolic model (GEMs). GEMs are a way of presenting Gene-Protein-Reaction (GPRs) occurring in an organism. There have been many studies on pathway analysis of bacteria through GEMs.8,9 However, no significant findings have been reported so far on archaea especially halophilic archaea except for the Halobacterium salinarum model.10,11 Since, H. pelagica CDK2 is well adapted to their niches and physical surroundings altering their nutritional capabilities and pathways the GEM of H. pelagica CDK2 will have its own significance.
The significant reduction in genome sequencing cost in last few years has resulted in an increase in the number of sequenced genomes all over the world. This has led to a rise in post genome sequencing technologies such as annotation, pathway analysis and genome scale metabolic models. In this study, we have generated a genome-scale metabolic model to allow pathway analysis of the organism. As we cannot completely rely on the accuracy of tools, GEMs often require manual intervention i.e., examining various databases and literature. This is by far the first study showcasing comprehensive pathway analysis of H. pelagica. We have also compared it with another H. pelagica genome isolated from China to find any correlation between the pathways and the geographical location.12
Acquisition of non-redundant genomic data
All the genomes of Halolamina species namely H. pelagica CDK2,6 H. pelagica CGMCC,12 H. rubra CBA1107,13 H. salifodinae DSM26232,14 H. sediminis halo7,15 Halolamina sp. R1-12, and Halolamina sp CBA1230 were downloaded from the NCBI genome database in the fasta format and were subjected to annotation using Prokkav1.12,16 Prokka was run with default parameters with a modification of “Kingdom: Archaea” as the subject database. All the genomes used with their respective accession numbers, genome size and number of coding sequences are presented in Table 1. The circular figure of the H. pelagica CDK2 genome was viewed using CGviewer17 for understanding the number of scaffolds, GC Content, GC skew+, and GC skew-.
Identification of H. pelagica CDK2 strain specific genes
The Prokka output (amino acid file) of each organism in faa format were input for Get_homologues by applying OMCL (Orthologues Markov Clustering)18,19 and COG (Cluster of orthologues)20 to obtain matrix containing number of gene families in number of organisms. Both COG and OMCL were used for clustering to get the high confidence clusters. The intersection of the two clusters is obtained through the perl script compare_clusters.pl. To find the strain-specific genes, the pangenome matrix is interrogated by the perl script parse_pangenome.pl.
Annotation of H. pelagica CDK2 by RAST and KEGG
Both the genome sequence of H. pelagica CDK2 and H. pelagica CGGHMS were subjected to annotation using RAST21 and KAAS.22 RAST was also implemented with default parameters mapping genes to different subsystems. KAAS provides functional annotation of genomes using manually curated groups of ortholog genes and automatically generate KEGG23 pathways. We also conducted Trnascan24 and Rnammer25 command line using default parameters with a kingdom as “Arc” to find transfer RNAs and ribosomal RNAs respectively.
Metabolic Reconstruction
A genome-scale metabolic model was constructed from the annotated genome of H. pelagica CDK2 using pathologic tool version 22.0 of the Pathologic database.26 It takes the annotated genome as input and connects the gene, protein to reactions using the Metacyc database. We manually curated the metabolic model with the following steps:
- Assign probable enzymes: These were the enzymes that the pathologic tool could not assign and left to the researcher to decide. Therefore, these enzymes were checked in both Rastk and Prokka annotation. Specific annotation is preferred over general annotation and non-hypothetical annotation is preferred over hypothetical protein.
- Carefully reviewing the ambiguous enzymes. An enzyme is said to be ambiguous if it is associated with multiple reactions.
Finally, the name matcher module of the pathologic tool was run and automatically filled the pathway holes.
Genomic features of Halolamina
The genome size and number of coding sequences of different Halolamina species ranged from 2.7 – 3.4 MB and 2808 – 3507, respectively. H. pelagica CDK2 has the highest number of coding sequences followed by Halolamina sp. CBA1230 (Table 1). The overall GC content of the class “Halobacteria” ranges from 44% to 70% but, specifically, the genome of H. pelagica CDK2 was found to have a high GC content of 67.6%. Along with H. pelagica CDK2, other species of genus Halolamina were also found to have similar GC content ranging from 66.73% to 69%. The circular figure of the H. pelagica CDK2 genome showing three scaffolds (red), GC Content (black), GC skew+ (green) and GC skew- (purple) is given in Fig. 1.
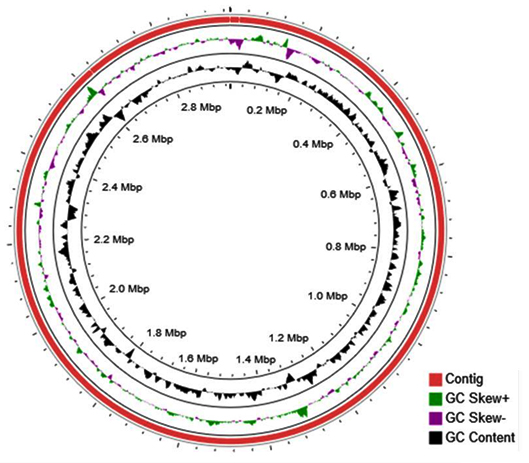
Fig. 1. Circular genome of Halolamina pelagica CDK2.
Analysis of Halolamina pelagica strain specific genes
The total gene clusters generated according to OMCL and COG with the help of Get_Homologues are shown in Fig. 2. Orthologous Markov Clustering resulted in 418 clusters whereas COG resulted in 536 clusters. Intersection of COG and OMCL produced 5573 clusters of gene families. The strain-specific genes which include the genes present in H. pelagica CDK2 but not in any other strain are listed in Supplementary Table 1. Worth mentioning here are thermostable carboxypeptidase and DNA repair MutS protein.
Table (1):
Genomic resources of different species of halophilic archaea Halolamina used for constructing Halolamina pan-genome.
Organism Name |
Accession number |
Genome Size |
No. Of CDS |
|---|---|---|---|
Halolamina pelagica CDK2 |
LGUC00000000 |
29,72,542 |
3483 |
Halolamina pelagica CGGHMS |
FOXI00000000 |
31,60,633 |
3184 |
Halolamina salifodinae DSM 26232 |
JAGGLC000000000 |
27,52,321 |
2808 |
Halolamina sediminis halo7 |
CVUA00000000 |
28,35,858 |
2881 |
Halolamina sp. R1-12 |
JAANTI000000000 |
31,61,503 |
3188 |
Halolamina sp CBA1230 |
CP054587 |
34,65,332 |
3570 |
Halolamina rubra CBA1107 |
BBJN00000000 |
29,55,001 |
3077 |

Fig. 2. Intersection of Orthologues Markov Clustering and Cluster of orthologues.
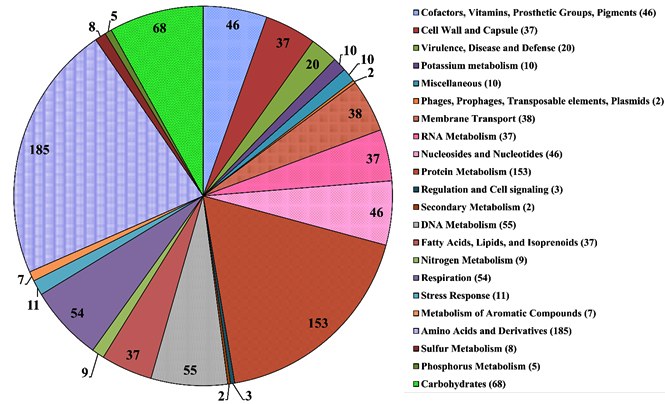
Fig. 3. Distribution of H. pelagica CDK2 genes in RAST Subsystem (numbers in parenthesis and pie-chart blocks are the subsystem feature counts).
RAST subsystems of H. pelagica CDK2 identifies plant growth-promoting (PGP) traits
Comparison of H. pelagica CDK2 and H. pelagica CGMCC in terms of ribosomal RNAs, transfer RNAs, RAST and Prokka annotation is given in Table 2. According to RAST, there was only 17% subsystem coverage corresponding to 174 subsystems for H. pelagica CDK2. The functional categories of RAST to which genes are assigned are represented as a pie chart in Fig. 3. The top five categories correspond to basic processes where highest proportion of genes (184) correspond to the subsystem “Amino acid and derivatives” followed by the subsystem “Protein metabolism” (154). The other three subsystems showing dominance are Carbohydrate metabolism (66), DNA metabolism (55) and Respiration (54). A detailed report of the RAST subsystem can be accessed through Supplementary Table 2. We found genes of phosphate metabolism like polyphosphate kinase, alkaline phosphatase, phosphate ABC transporters and phosphate transport system regulatory protein PhoU. Other important PGP traits were superoxide dismutase and catalase-peroxidase.27,28 A carbon starvation protein A corresponding to subsystem “carbon starvation” was also annotated.
Table (2):
Comparison of polished Halolamina pelagica CDK2 versus Halolamina pelagica CGGHMS.
CDK2 |
CGGHMS |
|
|---|---|---|
Size |
2972542 |
3160633 |
Number of Scaffolds |
3 |
37 |
Rast annotation (Number of CDS) |
3655 |
3458 |
Number of ribosomal RNA |
4 |
4 |
Number of transfer RNA |
47 |
42 |
Prokka annotation (Number of CDS) |
3483 |
3184 |
Number of misc. RNAs |
5 |
6 |
Number of ribosomal RNA |
6 |
5 |
Number of transfer RNA |
54 |
50 |
KEGG database analysis
Annotation against the KEGG database23 resulted in 44.3% (1377) coding sequences for H. pelagica CDK2 being assigned to KEGG pathways with the highest proportion of genes in Genetic Information and processing (213) followed by Protein families: Genetic Information processing (152). The other three top categories being Unclassified (132), Carbohydrate Metabolism (125) and Protein families: Signalling and cellular processes (117). Almost the same trend is observed for CGHMS strain except the fifth highest in CDK2 correspond to “Protein Families: Signalling and Cellular processes” (117) contrary to “Amino acid metabolism” (119) in CGGHMS. Fig. 4 shows the graphical comparison between the two strains in terms of KEGG pathways. A detailed report of genes and KEGG pathways for H. pelagica CDK2 is given in Supplementary Table 3. KEGG identifies bifunctional gene named trehalose synthase and phosphatase for trehalose biosynthesis. It showed myriad of metal resistant genes for copper, arsenic and fluoride. Other important proteins were catalase-peroxidase (KatG) and superoxide dismutase of “longevity regulating pathway”.
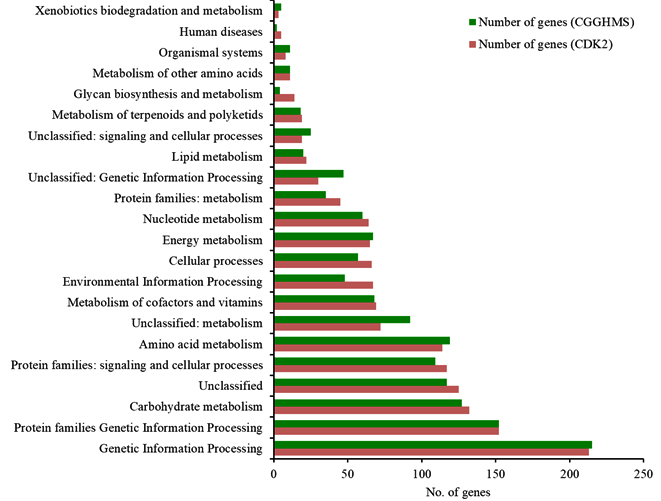
Fig. 4. KEGG pathway distribution and comparison of H. pelagica CDK2 and H. pelagica CGGHMS.
General features of Genome scale metabolic model of H. pelagica CDK2
The pathway analysis by Pathologic reported 105 pathways, 1170 enzymatic reactions, 8 transport reactions, 3662 polypeptides, 760 enzymes, 24 transporters, 1147 compounds and gene ontology (GO) terms assigned as 661. A metabolic model of H. pelagica CDK2 predicting GPR (gene-protein-reaction of pathways) is available at https://doi.org/10.5281/zenodo.5543458. Modified shikimate pathway is observed in the metabolic model of H. pelagica CDK2 credited to the presence of all the required enzymes such as 3-dehydroquinate dehydratase I (aroD), chorismate synthase (aroC), shikimate dehydrogenase (aroE), shikimate kinase (aroK) and 3-phosphoshikimate 1-carboxyvinyltransferase (aroA).29 Only two final enzymes phosphogluconate dehydrogenase and phosphoriboisomerase of oxidative pentose phosphate pathway were observed. Phosphomevolonate decarboxylase and isopentanyl kinase corresponding to modified mevolonate pathway were identified whereas enzymes phosphomevolonate kinase and diphosphomevolonate decarboxylase were not present.
Halophilic archaea have generated significant interest among microbiologists because of their characteristics to withstand a high salt environment, ability to endure high metal toxicity, high UV rays,2 and their existence in the extra-terrestrial environment.3 Among halophilic archaea, H. pelagica was first reported in 200930 and its strain CDK2 was sequenced in 2015.6 Its presence in the rhizosphere of plants growing in hypersaline soils of Rann of Kutch, Gujarat in all the seasons and its ability to produce plant growth-promoting attributes such as nutrient solubilisation and phytohormone production4,5 made it a worthy candidate for studying the underlying mechanisms and pathway analysis. Additionally, high variability among different halophilic archaea reveals a uniqueness associated with each of them.7 In the present study, we functionally analysed its genome and construct pathway analysis of the organism to uncover its resistant mechanisms which might have developed to endure abiotic stress such as high salinity, temperature and toxic metal.
H. pelagica CDK2 with a genome size of 2972152 bp has 67.6% GC content. High GC content is associated with high selective pressure in microbes and also contribute to the stability of the genome.31 The probable reason is the stacking energy of the GC which is higher than AT. This could also be the reason for this microbe being highly resilient to extreme environmental pressure (temperature, osmotic stress, and UV radiation). Furthermore, high GC content also leads to high energy efficient amino acids with differing amino acid usage pattern, making H. pelagica endure a harsh environment by conserving energy.
Other than H. pelagica CDK2, H. pelagica CGMCC was also isolated from the hypersaline regime, Taibei marine solar saltern.12 We compared the genome sequences of these two Halolamina strains to find if there is any correlation between the gene features and geographical location. Cui et al.30 studied H. pelagica (TBN49 and TBN21) isolated from the same geographic location and still found the difference in the assimilation of nitrate to nitrite, where TBN21 was unable to assimilate nitrate and nitrite N2, obviously reflecting the differences of pathways.30 These differences among strains of H. pelagica (TBN49 and TBN21) despite of isolation from same geographical location, points out the importance of comparing the genome sequence of CDK2 and CGMCC strains of the same species but isolated from different geographical locations and temperature regimes. The Rann of Kutch is dominated by high temperatures whereas Taibei marine saltern has a medium temperature of 25°C. We found genes exclusively present in H. pelagica CDK2 in comparison to H. pelagica CGMCC corresponding to the high temperature tolerance (Supplementary File 1). Small heat shock protein 16.532 protect the proteins from thermal denaturation and therefore allows proper functioning of the cell. Similarly, DNA repair protein MutS repairs nicked DNA which is induced by UV rays.33 Last but not the least, thermostable carboxypeptidase was also present. Carboxypeptidases catalyses the C-terminal hydrolysis (hydrolysis of peptide bond at C-terminal) of proteins and peptides majorly, proteins containing neutral, aromatic, basic and polar amino acids.34
As stated earlier, H. pelagica CDK2’s presence in the rhizosphere of wild vegetation and positive confirmation of phosphorus solubilisation test describe the need to investigate the occurrence of genes for the PGP traits. We found ABC transporters for phosphorus uptake (pho), pyrophosphate kinase (ppk), and alkaline phosphatase (alp). The basic physiological function of alkaline phosphatase is dephosphorylation and this enzyme is found across the domains.35,36 It is a periplasmic enzyme, heat stable and involved in supplying inorganic phosphate to the cells in phosphate limiting environments through dephosphorylation of organic compound.37 The inorganic phosphate made available is then transported across the cell membrane employing phosphate specific transport systems which might involve phosphate ABC transporter. ppk is also one of the strain-specific genes present in H. pelagica CDK2 and no significant reports are available in the literature that corroborates the presence ppk gene in other species of Halolamina currently present in NCBI. Several genes responsible for nullifying reactive oxygen species produced excessively in stressed environments were annotated like superoxide dismutase and catalase-peroxidases. There are reports which indicated, using these antioxidant enzymes produced by microbes helps plants as well to cope with the extreme conditions.28,29 Nitrous oxide reductase responsible for reducing greenhouse gas into dinitrogen was also annotated in the genome. Worth mentioning is that nitrous oxide has a 300-fold greater ability to deplete the ozone layer than CO2.
Earlier halophilic archaea belonging to class Halobacteria were known to only employ salt in strategy that is the accumulation of KCl in the cell in response to the high salt environment. But this notion has changed with the discovery of genes related to trehalose biosynthesis and organic solutes accumulation which is also called the “low salt in, organic solutes” strategy. On investigation, we also found bifunctional protein incorporating trehalose 6 phosphate synthase and phosphatase activity. This enzyme leads to the synthesis of trehalose which is a well-known compatible solute. There has been experimental evidence of the significance of compatible solutes for microorganisms that are flexible and could grow at a lower salt concentration as well.38 In our laboratory, we could grow the culture of the strain at a minimum of 50-80 gL-1 of salt concentration and maximum up to 250 gL-1 concentrations which are wide adaptability of salt concentration again pointing to its ability to accumulate compatible solutes (Unpublished report). Furthermore, three prominent symporters neurotransmitter/sodium symporter (NSS), solute/sodium symporter and proline/sodium symporter (SSS) were also found in CDK2. putP (proline/sodium symporter) gene product can accumulate proline and help the cell to grow in high osmotic stress.39 In Bacillus subtilis, the gene is upregulated by sigma A and B promoters which are stress-induced promoters. NSS family, which is required to transport osmolytes, amino acids, neurotransmitters were also identified but the subfamily could not be found. Monovalent Na+/proton antiporters were also annotated which are a complex of seven proteins working in a harmony to get rid of Na+ and maintain the cell’s pH.40 These are divided into three groups based on the rearrangement of genes in an operon. Accordingly, the operon present in H. pelagica CDK2 (DDDCBBGFE) belongs to “group3/ Additional group” as it has a missing mrpA gene and duplication of mrpB, mrpC and mrpD. Halobacterium salinarium has the same operon “DDDCBBGFE” whereas Haloarcula marismortui ATCC 43019 has “EFGBBCDDD”,41 The significance of gene order in an operon is still an open question.
As previously mentioned, the Rann of Kutch is a solar saltern that has a high concentration of salt due to evaporation of water and this also concentrates metal ions present in the soil. Heavy metals like Copper, Zinc, Iron, Manganese, Nickel and Cobalt are good in nano-molar to micro-molar concentrations but are harmful at higher concentrations, resulting in cell death. Therefore, microorganisms living in this region must have developed mechanisms to resist or get rid of the toxic effects of metals and Halolamina pelagica is no different as witnessed by the myriad of metal efflux and resistant genes. The main mechanism of carrying out copper efflux is through copper transporting ATPases.42 CopA and CopB which are P-type ATPase transporters known for transporting Cu(2) and Cu(3) respectively were annotated in the genome. CopZ which is a copper-binding protein and metalloprotease was also found. However, we could not find CopY in the H. pelagica genome. Genes for arsenic resistance were also found in H. pelagica CDK2 such as arsA, arsB, arsC and arsR. arcC is an arsenate reductase that converts arsenate to arsenite and then is transported out of the cell by the action of arsA (Arsenite translocating ATPases) and arsB (transporter).43 The regulation is maintained by arsR which in the absence of arsenite, represses the ars operon. The arsR repression is also seen in other archaea which dwell in a high arsenic environment but lack gene arsC. Therefore, many researchers have pointed out the different mechanisms of arsR resistance in archaea. However, H. pelagica CDK2 have all the genes except arsD. crcB gene which is considered a fluoride exporter is also annotated in the genome. These are highly selective ion channels that do not transport even very similar chloride ions, preventing the harmful concentration of fluoride inside the cell as fluoride is known to inhibit many important processes such as glycolysis and polymerization.44
An enzyme (AOR) aldehyde oxidoreductase is also identified in H. pelagica which requires specifically tungsten for its activity and synthesis. Therefore, we investigated the presence of tungsten uptake system and found all the genes (tupA, tupB and tupC)45 in an operon responsible for highly specific tungsten uptake contrary to the previous research by Zhang and Gladyshev, where they suggest wtp system is widespread in archaea for tungsten and molybdenum uptake. However, moco biosynthesis genes46 are also present in H. pelagica and it is a known fact that tungsten and molybdate are chemically very similar and additionally we could not find a molybdenum (modABC) uptake system in H. pelagica. Therefore, we hypothesized that tupABC might also be responsible for molybdenum uptake when required by the organism for moco biosynthesis.
Nickel and Cobalt are essential elements required as a cofactor for many enzymes but present in very low quantity in the environment. Many enzymes like urease, Ni-Fe hydrogenases, carbon monoxide dehydrogenases contain nickel as an essential cofactor. Surprisingly, we could not find any specific uptake system like nikABC for nickel in H. pelagica. Though we could find CbiMNOQ system for cobalt uptake which can process nickel uptake as well. cbiMN acts as a cobalt transporter whereas cbiO is involved in releasing energy with ATP hydrolysis for the uptake of cobalt and nickel.47 Additionally, nikR which is a nickel responsive regulator is also present upstream of cbiMNOQ indicating its role in nickel transport as well. It will not be wrong to say this is the reason for H. pelagica CDK2 concise genome and a way of conserving energy by using one complex for two metal transport (Cobalt/nickel and Molybdenum/tungsten). corA gene is also identified in H. pelagica CDK2 which helps the cell to uptake magnesium from the surroundings.48 ZnuABC is a highly specific group of ABC transporters responsible for zinc uptake from the environment.49 There are many reports of its presence in bacteria but to the best of our knowledge, no significant reports exist of ZnuABC for halophilic archaea instead ZupT transporters are widespread in archaea.50 However, our findings suggest that ZnuABC operon works in CDK2 as a zinc uptake system. There are different genes associated with the efflux of copper in different archaea.
According to the genome-scale metabolic model, biosynthesis of all amino acids except serine and methionine are reported in H. pelagica CDK2. However, we could grow H. pelagica CDK2 under in vitro conditions in the absence of any amino acid in growth media. We could not find a classical shikimate pathway that enrols erythrose-4 -phosphate and phospho-enol-pyruvate to produce chorismate through all enzymes of a pathway from chorismate to tryptophan biosynthesis in H. pelagica CDK2. It has been proposed for the archaeon Methanocaldococcus janneschii that an alternate pathway exists requiring the condensation of 4-semialdehyde and 6-deoxy-5-ketofructose to form dihydroshikimate which in turn converts to shikimate.29 The same has been proposed for Natronomonas pharaonis.51 We looked for the same enzymes in H. pelagica CDK2 and found all the enzymes namely aroD 3-dehydroquinate dehydratase I, aroC chorismate synthase, aroE shikimate dehydrogenase, aroK shikimate kinase, aroA 3-phosphoshikimate 1-carboxyvinyltransferase and aroC. Therefore, a possibility of a similar pathway in H. pelagica CDK2 can’t be ruled out.
For the synthesis of membrane lipids and other isoprenoids, the modified MVA pathway occurs in archaea as discovered by Grochowski et al.52 This modified pathway employs phosphomevalonate decarboxylase (PMD) for the conversion of isopentanyl phosphate (IP) from MVA5P. Interestingly, this gene is known to be conserved in halophilic archaea53 and H. pelagica CDK2 too represented all the enzymes required for the modified mevalonate pathway including PMD.
Similar to Halobacterium salinarum, we could identify final enzymes of OPPP (oxidative branch of pentose phosphate pathway) namely phosphogluconate dehydrogenase and phosphoriboisomerase whereas reverse ribulose monophosphate pathway and non-oxidative branch of pentose phosphate pathway are lacking. Orland Gonzalez has connected the initial steps of the semiphosphorylated ED pathway to the final steps of OPPP.10,54 If this connection is true, the same connection cannot be ruled out in H. pelagica CDK2.
Comprehensive genome analysis of halophilic archaea, H. pelagica CDK2 revealed genes such as alkaline phosphatase and pyrophosphokinase involved in P solubilisation coinciding with our previous research.45 Strain-specific attributes of H. pelagica CDK2 like thermostable carboxypeptidase and DNA repair protein MutS required for the integrity of protein and DNA respectively from high-temperature denaturation is also reported. A possession of a bifunctional enzyme trehalose synthase/phosphatase responsible for trehalose biosynthesis indicated wide adaptability of H. pelagica CDK2 to varying salt concentrations. Previous reports has suggested the accumulation of organic solutes for wide adaptability as opposed to the accumulation of KCl for narrow adaptability.38 Our study has also pointed out H. pelagica CDK2’s clever use of one ABC transporter for two similar metals like that of Nickel/cobalt and Molybdenum/Tungsten probably to conserve energy. This is the first study that has comprehensively deciphered the genomic features and probable salt strategies employed by H. pelagica which can be researched further for transfer in crops. The metabolic model constructed can be utilized for flux balance studies where its nutritional requirements can be extensively studied. The metabolic model can also be integrated with transcriptomic studies with varying salt concentrations to develop a constraint-based metabolic model.
Additional file: Additional Table 1.
ACKNOWLEDGMENTS
The authors would like to thank Division of Microbiology, ICAR-Indian Agricultural Research Institute, New Delhi, India for providing the facilities.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
AUTHORS’ CONTRIBUTION
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
FUNDING
The study was financially supported by the Department of Biotechnology, Ministry of Science and Technology, Government of India through grant number: BT/PR6540/BCE/8/917/2012
ETHICS STATEMENT
This article does not contain any studies with human participants or animals performed by any of the authors.
AVAILABILITY OF DATA
All datasets generated or analysed during this study are included in the manuscript and available as supplementary tables and files
- Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci U S A. 1990;87(12):4576-4579.
Crossref - Grant WD, Gemmell RT, McGenity TJ. Halobacteria: The evidence for longevity. Extremophiles. 1998;2(3):279-287.
Crossref - Treiman AH, Gleason JD, Bogard DD. The SNC meteorites are from Mars. Planet Space Sci. 2000;48(12):1213-1230.
Crossref - Yadav AN, Sharma D, Gulati S, et al. Haloarchaea Endowed with Phosphorus Solubilization Attribute Implicated in Phosphorus Cycle. Sci Rep. 2015;5:12293.
Crossref - Yadav AN, Gulati S, Sharma D, et al. Seasonal variations in culturable archaea and their plant growth promoting attributes to predict their role in establishment of vegetation in Rann of Kutch. Biologia. 2019;74(8):1031-1043.
Crossref - Gaba S, Singh RN, Abrol S, Yadav AN, Saxena AK, Kaushik R. Draft Genome Sequence of Halolamina. pelagica CDK2 from natural salterns of Rann of Kutch, Gujarat, India. Genome Announcements. 2017;5(6):e01593-16.
Crossref - Gaba S, Kumari A, Medema M, Kaushik R. Pan-genome analysis and ancestral state reconstruction of class halobacteria: probability of a new super-order. Sci Rep. 2020;10(1):21205.
Crossref - Mandakovic D, Cintolesi A, Maldonado J, et al. Genome-scale metabolic models of Microbacterium species isolated from a high altitude desert environment. Sci Rep. 2020;10(1):5560.
Crossref - Gomez-Garzon C, Hernandez-Santana A, Dussan J. A genome-scale metabolic reconstruction of Lysinibacillus sphaericus unveils unexploited biotechnological potentials. PLoS One. 2017;12(6):1-21.
Crossref - Gonzalez O, Gronau S, Pfeiffer F, Mendoza E, Zimmer R, Oesterhelt D. Systems Analysis of Bioenergetics and Growth of the Extreme Halophile Halobacterium salinarum. PLoS Comput Biol. 2009;5(4):e1000332.
Crossref - Gonzalez O, Gronau S, Falb M, et al. Reconstruction, modeling & analysis of Halobacterium salinarum R-1 metabolism. Mol Biosyst. 2008;4(2):148-159.
Crossref - Whitman WB, Woyke T, Klenk HP, et al. Genomic encyclopedia of bacterial and archaeal type strains, phase III: The genomes of soil and plant-associated and newly described type strains. Stand Genomic Sci. 2015;10:26.
Crossref - Cha IT, Yim KJ, Song HS, et al. Halolamina rubra sp. nov., a haloarchaeon isolated from non-purified solar salt. Antonie van Leeuwenhoek. 2014;105(5):907-914.
Crossref - Zhang WY, Huo YY, Zhang XQ, Zhu XF, Wu M. Halolamina salifodinae sp. nov. and Halolamina salina sp. nov., two extremely halophilic archaea isolated from a salt mine. Int J Syst Evol Microbiol. 2013;63:4380-4385.
Crossref - Koh HW, Song HS, Song U, Yim KJ, Roh SW, Park SJ. Halolamina Sediminis sp. nov., an extremely halophilic archaeon isolated from solar salt. Int J Syst Evol Microbiol. 2015;65(8):2479-2484.
Crossref - Hyatt D, Chen G-L, LoCascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010;11(1):119.
Crossref - Grant JR, Stothard P. The CGView Server: a comparative genomics tool for circular genomes. Nucleic Acids Res. 2008;36(Suppl 2):181-184.
Crossref - Contreras-Moreira B, Vinuesa P. GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl Environ Microbiol. 2013;79(24):7696-7701.
Crossref - Li L, Stoeckert CJJ, Roos DS. OrthoMCL: Identification of Ortholog Groups for Eukaryotic Genomes. Genome Res. 2003;13(9):2178-2189.
Crossref - Tatusov RL, Galperin MY, Natale DA, Koonin E V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000;28(1):33-36.
Crossref - Aziz RK, Bartels D, Best A, et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genomics. 2008;9:75.
Crossref - Kanehisa M, Sato Y, Morishima K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J Mol Biol. 2016;428(4):726-731.
Crossref - Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 1999;27(1):29-34.
Crossref - Chan PP, Lowe TM. tRNAscan-SE: Searching for tRNA genes in genomic sequences. Methods Mol Biol. 2019;1962(5):1-14.
Crossref - Lagesen K, Hallin P, Rødland EA, Stærfeldt HH, Rognes T, Ussery DW. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35(9):3100-3108.
Crossref - Karp PD, Paley S, Romero P. The pathway tools software. Bioinformatics. 2002;18(SUPPL. 1):225-232.
Crossref - Leontidou K, Genitsaris S, Papadopoulou A, et al. Plant growth promoting rhizobacteria isolated from halophytes and drought-tolerant plants: genomic characterisation and exploration of phyto-beneficial traits. Sci Rep. 2020;10(1):14857.
Crossref - Bruto M, Prigent-Combaret C, Muller D, Moenne-Loccoz Y. Analysis of genes contributing to plant-beneficial functions in plant growth-promoting rhizobacteria and related Proteobacteria. Sci Rep. 2014;4:6261.
Crossref - White RH. L-aspartate semialdehyde and a 6-deoxy-5-ketohexose 1-phosphate are the precursors to the aromatic amino acids in Methanocaldococcus jannaschii. Biochemistry. 2004;43(23):7618-7627.
Crossref - Cui HL, Gao X, Yang X, Xu XW. Halolamina pelagica gen. nov., sp. nov., a new member of the family Halobacteriaceae. Int J Syst Evol Microbiol. 2011;61(7):1617-1621.
Crossref - Raghavan R, Kelkar YD, Ochman H. A selective force favoring increased G+C content in bacterial genes. Proc Natl Acad Sci U S A. 2012;109(36):14504-14507.
Crossref - Kim R, Kim KK, Yokota H, Kim SH. Small heat shock protein of Methanococcus jannaschii, a hyperthermophile. Proc Natl Acad Sci U S A. 1998;95(16):9129-9133.
Crossref - Acharya S, Foster PL, Brooks P, Fishel R. The coordinated functions of the E. coli MutS and MutL proteins in mismatch repair. Mol Cell. 2003;12(1):233-246.
Crossref - Matsumura E, Sato T, Toyoda N. Isolation and characterization of a microbial Arg/Lys carboxypeptidase, carboxypeptidase F. Lett Appl Microbiol. 1995;20(3):157-159.
Crossref - Qureshi AS, Marium S, Khushk I, Bhutto MA. Production of alkaline phosphatase from newly isolated Aspergillus fumigatus EFRL05. Topclass J Microbiol. 2013;1(4):67-73.
- Kumar M, Kaur PP, Ganjewala D. Isolation of periplasmic alkaline phosphatase from Rhizobium bacteria. Res J Microbiol. 2008;3:157-162.
Crossref - Qureshi A, Dahot M, Panhwar S. Biosysnthesis of alkaline phosphatase by Escherichia coli efrl 13 in submerged fermentation. World Appl Sci J. 2010;8:50-56.
- Youssef NH, Savage-Ashlock KN, McCully AL, et al. Trehalose/2-sulfotrehalose biosynthesis and glycine-betaine uptake are widely spread mechanisms for osmoadaptation in the Halobacteriales. ISME J. 2014;8(3):636-649.
Crossref - Henriquez T, Wirtz L, Su D, Jung H. Prokaryotic solute/sodium symporters: Versatile functions and mechanisms of a transporter family†. Int J Mol Sci. 2021;22(4):1880.
Crossref - Fukaya F, Promden W, Hibino T, Tanaka Y, Nakamura T, Takabe T. An Mrp-like cluster in the halotolerant cyanobacterium Aphanothece halophytica functions as a Na+/H+ antiporter. Appl Environ Microbiol. 2009;75(20):6626-6629.
Crossref - Swartz TH, Ikewada S, Ishikawa O, Ito M, Krulwich TA. The Mrp system: A giant among monovalent cation/proton antiporters? Extremophiles. 2005;9(5):345-354.
Crossref - Ettema TJG, Brinkman AB, Lamers PP, Kornet NG, de Vos WM, van der Oost J. Molecular characterization of a conserved archaeal copper resistance (cop) gene cluster and its copper-responsive regulator in Sulfolobus solfataricus P2. Microbiology. 2006;152(7):1969-1979.
Crossref - Voica DM, Bartha L, Banciu HL, Oren A. Heavy metal resistance in halophilic Bacteria and Archaea. FEMS Microbiol Lett. 2016;363(14):1-9.
Crossref - Stockbridge RB, Robertson JL, Kolmakova-Partensky L, Miller C. A family of fluoride-specific ion channels with dual-topology architecture. Elife. 2013;2:1-14.
Crossref - Makdessi K, Andreesen JR, Pich A. Tungstate Uptake by a Highly Specific ABC Transporter in Eubacterium acidaminophilum. J Biol Chem. 2001;276(27):24557-24564.
Crossref - Mendel RR. The molybdenum cofactor. J Biol Chem. 2013;288(19):13165-13172.
Crossref - Rodionov DA, Hebbeln P, Gelfand MS, Eitinger T. Comparative and functional genomic analysis of prokaryotic nickel and cobalt uptake transporters: Evidence for a novel group of ATP-binding cassette transporters. J Bacteriol. 2006;188(1):317-327.
Crossref - Kehres DG, Lawyer CH, Maguire ME. The CorA magnesium transporter gene family. Microb Comp Genomics. 1998;3(3):151-169.
Crossref - Patzer SI, Hantke K. The ZnuABC high-affinity zinc uptake system and its regulator Zur in Escherichia coli. Mol Microbiol. 1998;28(6):1199-1210.
Crossref - Zhang T, Liu J, Fellner M, Zhang C, Sui D, Hu J. Crystal structures of a ZIP zinc transporter reveal a binuclear metal center in the transport pathway. Sci Adv. 2017;3(8):1-9.
Crossref - Gonzalez O, Oberwinkler T, Mansueto L, et al. Characterization of growth and metabolism of the haloalkaliphile Natronomonas pharaonis. PLoS Comput Biol. 2010;6(6):e1000799.
Crossref - Grochowski LL, Xu H, White RH. Methanocaldococcus jannaschii uses a modified mevalonate pathway for biosynthesis of isopentenyl diphosphate. J Bacteriol. 2006;188(9):3192-3198.
Crossref - Hayakawa H, Motoyama K, Sobue F, et al. Modified mevalonate pathway of the archaeon Aeropyrum pernix proceeds via trans-anhydromevalonate 5-phosphate. Proc Natl Acad Sci U S A. 2018;115(40):10034-10039.
Crossref - Kennedy SP, Ng WV, Salzberg SL, Hood L, DasSarma S. Understanding the adaptation of Halobacterium species NRC-1 to its extreme environment through computational analysis of its genome sequence. Genome Res. 2001;11(10):1641-1650.
Crossref
© The Author(s) 2022. Open Access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License which permits unrestricted use, sharing, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.