


ISSN: 0973-7510
E-ISSN: 2581-690X
Several reports on genome sequencing using Next Generation Sequencing (NGS) to identify the total genome were extensively carried out; however, genome identification on thermophilic microorganisms is still limited. In this report, genome identification of thermophilic microorganisms isolated from compost, namely Pseudoxanthomonas taiwanensis AL17, was carried out. The result showed that AL17 contains 3,064,463 bp with a GC content of 72.08%. The genome comprises 2,833 CDSs, 6 RNA (5S, 16S, and 23S), 48 tRNA genes, and 17 Pseudogenes. A comparison of the genome to data based on Average Nucleotide Identity from Dfast-qc shows that the genome is closely related to Pseudoxanthomonas taiwanensis. Further analysis discovers numerous genes coding for potential enzymes, including hydratase, transferase, dehydrogenase, exopeptidase and hydrolases. In addition, the genome exhibits a number of stress-tolerant genes. Detailed analysis of the hydrolase genes, especially for esterase and lipase, showed that the genome exhibits no true lipase but a lipolytic enzyme within the GDSL-type esterase/lipase motif. The genomic information provides an understanding of thermophilic genomes and their relevance to stress-tolerant adaptation and explores potential genes, especially for industrial applications.
Compost, Genome, Pseudoxanthomonas taiwanensis, Thermophilic
Microorganisms based on temperature growth were categorized into three types, psychrophilic (-3-20°C), mesophilic (13-45°C), and thermophilic (≥42°C or more).1 Enzymes derived from thermophilic microorganisms exhibit many advantages, including expressing various valuable metabolite pathways, better catalytic activity, and more stable enzymes in extreme environment.2 The discovery of enzymes derived from thermophilic microorganisms was widely used to develop biotechnological processes.
Compost is one source of thermophilic microorganisms.3-5 P. taiwanensis AL17 was isolated from household compost at Saraga ITB Bandung.5 Phylogenetic analysis based on the 16S rRNA gene showed that the isolate was closely related to P. taiwanensis NBRC 101072 (99%). The organism is a typical thermophilic bacterium that expresses many industrial enzymes.5,6 The bacterium is a gram-negative, aerobic, thermophilic, unform spores,6 expressing potential metabolites for commercial application, including hydrolases such as amylase, cellulase, and lipase.7 Recently reports on the presence of various unique microbial populations in thermogenic environments were published worldwide,8 such as microbial analysis from biomass waste from Turkey,9 hot spring in China,10 food waste compost in Malaysia,11 textile waste compost in Lithuania,12 and Cangar hot spring in Malang, Indonesia.13 The use of NGS techniques in the microbial community is becoming increasingly developed. It allows the conducting of rapid, efficient, and accurate studies on the genetics of microorganisms and functional diversity in extreme environments.14,15
Utilization of NGS to detect the presence of strict anaerobic, chemolithotrophic, and acidothermophilic bacteria was reported.8 The NGS analysis uncovered hydrocarbon-degrading thermophiles and essential metabolic pathways for bacterial survival in harsh environments,14,16 and deeper insights into microbial ecology and their potential to support life in extreme environments.14 A previous study on P. taiwanensis AL17 showed the strain expressing some potential enzymes for industrial biocatalyst.5 However, cloning for true lipase from the organism was always unsuccessful. Therefore, genomic sequencing and subsequent prospecting of gene analysis were carried out to probe the potential genes from the isolate.
This study would like to probe and identify the metabolic potential genes of P. taiwanensis AL17 through a genomic approach. The availability of genomic data is highly relevant for the cloning approach. Genomic information may also explain the understanding of molecular adaptive mechanisms of microbes for survival in extreme environments.
Growth of Isolate AL17
Bacterial culture was provided from a collection of thermophilic microorganisms isolated from household and dry leaves compost at the Waste Disposal Site in Saraga, Institute Technology of Bandung.5 The glycerol stock was incubated in liquid media at 55°C overnight. Subsequently, the culture was transferred to fresh liquid media containing yeast extract (0.5% w/v), meat extract (0.5% w/v), NaCl (1% w/v), CaCl2 (1% w/v) then incubated at 55°C for 17 hours.
Genome sequencing and assembly
The Whole Genome Sequencing analysis was performed by Genetica Science Indonesia using the NGS method with the Oxford Nanopore Platform operated by MinKNOW software. Base calling was performed using Guppy with high accuracy mode.17 The quality of reading was visualized with NanoPlot.18
Genomic data assessment of AL17 was performed with Flye.19 The closely related species of the assembled sequence were identified using CheckM of dfast-qc.20 Mapping was performed with minimap2.21 The assembled sequence was polished four times by Racon and the first polished round by Medaka.22 The quality of the assembled sequence was determined using Quast.23 Annotation was performed using PGAP.24 Genome visualization was built by Circos.23
Genome properties
Identification of enzymes from the sequence used Rapid Annotation Subsystem Technology (RAST).25 FastANI version 0.1.326 was used to calculate ANI (Average Nucleotide Identity) while GGDC (Genome-to-Genome Distance Calculator) version 3.027 was used to calculate dDDH. A phylogenomic tree was created from the data set in OrthoFinder version 2.5.428 and visualized with iTOL version 6.5.2.29 The IslandViewer web server version 4 was used to predict the existence of a genomic island.30
Thermophilicity of P. taiwanensis AL17
Compost is a good source for novel mesophilic, thermophilic bacteria and fungi, particularly for biomass degradation.31 Along with end products such as humus, microorganisms in compost convert organic material into carbon dioxide, biomass, and heat energy.32 Therefore, the isolation of microorganisms from compost is of high interest. The enzymes from the organisms may applied for several potential commercial products, such as biodegradation and production of bioactive compounds, such as antibiotics and enzymes.31 P. taiwanensis AL17 was isolated from the thermogenic phase of the composting process.5 The organism exhibits optimum growth at 55°C.
The complete genome of P. taiwanensis AL17 was sequenced based on the NGS method. Annotating of the genome exhibits the presence of 2,833 total genes, with 2,775 protein-coding sequences (CDS). Furthermore, tRNA, rRNA, and ncRNA genes were identified (Table 1). In addition, pseudogenes and CRISPR arrays were detected. The presence of genome annotations was visualized using Circos software (Figure 1). The genome size of P. taiwanensis AL17 is approximately 3.1 Mb, with the GC composition reaching 72%, suggesting that P. taiwanensis AL17 is a thermophilic bacterium. A similar study conducted by Verma et al.33 on Thermobispora bispora was found to have a size of 4.1 Mb with an average GC content of 72.3% and Thermaerobacter subterranus showed a GC content of 72%. However, other reports indicated a low GC composition in some thermophilic and hyperthermophilic bacteria such as Thermotoga maritima, with a median genome size of 1.87 Mb and a GC content of 46.2% was reported 10. For Dictyoglomus sp., an overall GC content of approximately 33% was reported, with the median total genome length ranging from 1.6918 to 1.9569 Mb.34 The findings indicate that GC composition is not a major factor in the thermophilicity or hyperthermophilicity of bacteria. The optimum growth temperature of Aquifex aeolicus was at 95°C despite its low GC content of 43.4%.35 A correlation of genome size with the optimum growth temperature of thermophilic and hyperthermophilic bacteria was also inappropriate. Most bacteria classified as mesophiles exhibit larger genome sizes (above 6 Mb) compared to thermophiles and hyperthermophiles which tend to have shorter genome sizes, typically less than 3 Mb.36
Table (1):
Genome features of P. taiwanensis AL17
Genome size |
3,064,463 |
G + C content |
72.08% |
Completeness |
94.39% |
Contamination |
1.35% |
Total of genes |
2,833 |
Total protein-coding sequence (CDS) |
2,775 |
rRNA gene |
6 |
tRNA gene |
48 |
ncRNA |
4 |
Total pseudogenes |
170 |
CRISPR arrays |
2 |
Source |
Compost |
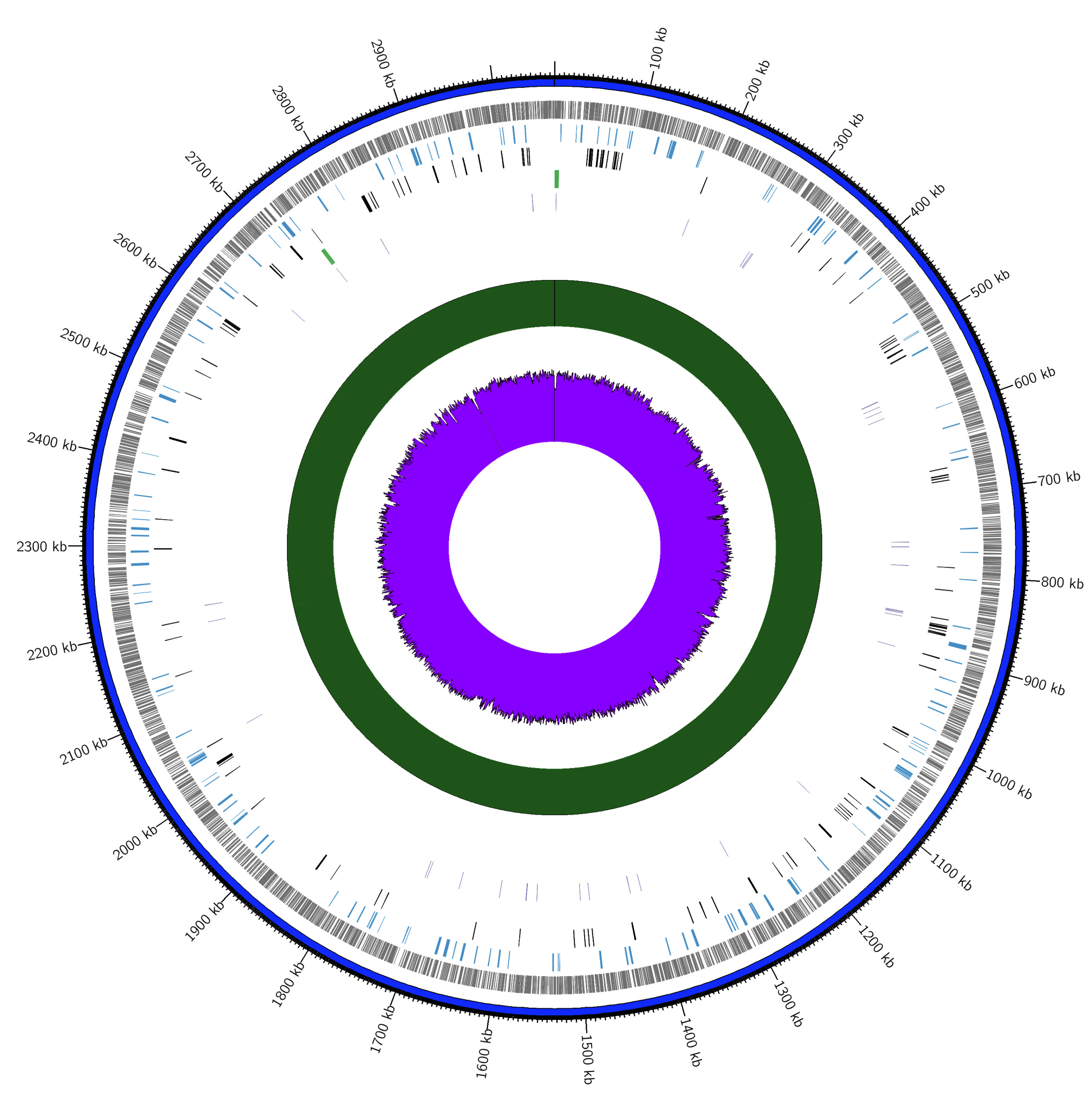
Figure 1. Genome visualization by Circos; contig (blue), genes (grey), pseudogenes (blue), CDS (black), rRNA (green), tRNA (purple), depth (depth >50 = green; depth <50 = red), GC content (GC content >50% = purple; GC content <50% = brown)
Several studies propose that mechanisms possessed by thermophilic bacteria with higher GC composition were to maintain the integrity of double-stranded DNA structure at high temperatures.37 However, the thermophilicity is not only determined by GC content but rather complex mechanisms. The other possibilities of thermophilic bacteria adapting at high temperatures are the presence of specific thermostable enzymes, such as reverse gyrase, assisting in replication and regulation of the genes at high temperatures38-40 or the presence of specific nucleotides which may contribute to thermal stability.41 The presence of nonspecific DNA-binding proteins like Smj12, topoisomerase I, and chaperone proteins DnaK, DnaJ, and GrpE play roles in positive DNA supercoiling.38 High purine and pyrimidine composition were also considered to provide low DNA flexibility and confer thermal resistance.37 Additionally, transient binding or association of histone and non-histone proteins with DNA may provide additional thermal resistance.42-45
Stress Tolerant of P. taiwanensis AL17
Average Nucleotide Identity (ANI) from Dfast-qc 20 was used to compare the P. taiwanensis AL17 genome with others. The result showed that P. taiwanensis AL17 was closely related to Pseudoxanthomonas taiwanensis DSM22914 (99.03%). This is in agreement with previous data, based on 16S rRNA gene.5 In addition to high thermophilicity, the isolate showed methanol tolerance.5
P. taiwanensis is a thermophilic bacteria associated with stress tolerance such as high temperatures, extreme pH levels, and toxic compounds.46 The genome sequence of P. taiwanensis AL17 revealed several genes related to stress tolerance such as osmotic, oxidative, heat-shock stresses, alkaline resistance, and carbon starvation (Table 2). In addition, the genome also contains the spermidine synthase gene, probably responsible for the import of the spermidine/putrescine ABC transporters and related to the thermophilicity of the isolate.47,48 The genome of Geobacillus kaustophilus was reported to exhibit unique genes, including spermine/spermidine synthase and polyamine ABC transporter (permease), which were responsible for the thermophilicity of the species.49
The genes associated with heat-shock protein were also found on P. taiwanensis AL17 including GrpE, DnaK, DnaJ, GroEL chaperones, GroES co-chaperone, and heat-inducible transcription repressors (Table 2). The data agrees with the genome of Geobacillus sp. isolate WSUCF1.50 Other genes involved in the thermophilic environment coding for PriA helicase and the DNA-binding protein HU.51 Several genes associated with the thermophilic environment including PriA helicase, DNA-binding protein HU, GrpE, DnaK, DnaJ, GroEL chaperones, GroES co-chaperone, and heat-inducible transcription repressors were revealed on the P. taiwanensis AL17 genome (Table 2). The activity of some proteins including the molecular chaperones GroEL, GroES, HrcA, GrpE, and DNA-binding proteins like RuvB, RecA, and RecG was found to play a significant role in maintaining cellular functions under heat stress.52,53 Moreover, two genes encoding the cold-shock protein, namely the CsPA gene, were found in the genome of P. taiwanensis AL17 (Table 2). In addition, several genes were found in the isolate (Table 2) to express proteins resistant to osmotic stress, such as the OpuD osmoprotectant-related transporter (glycine betaine transporter) and the Na+/H+ antiporter coding genes, NhaA and NhaD, related to alkaline pH adaptation. P. taiwanensis AL17 genome also contains a gene that codes for sulfate permease to adapt to high sulfate environments. This is similar to the genome of Geobacillus sp. WSUCF1 consisting several genes for pH adaptation.50
Table (2):
Thermal stress related to genes of P. taiwanensis AL17 genome
| Feature ID | Genes |
|---|---|
| Heat adaptation | DNA gyrase subunit A (EC 5.99.1.3) |
| DNA gyrase subunit B (EC 5.99.1.3) | |
| S-adenosylmethionine decarboxylase proenzyme (EC 4.1.1.50), prokaryotic class 1A | |
| DNA-binding protein HU | |
| Spermidine synthase (EC 2.5.1.16) | |
| Arginine decarboxylase (EC 4.1.1.19). | |
| Heat responses | tmRNA-binding protein SmpB |
| Translation elongation factor LepA | |
| Heat-inducible transcription repressor HrcA | |
| Heat shock protein GrpE | |
| Chaperone protein DnaK | |
| Chaperone protein DnaJ | |
| Ribosomal protein L11 methyltransferase (EC 2.1.1.-) | |
| Ribosome-associated heat shock protein implicated in the recycling of the 50S subunit (S4 paralog) | |
| Ribonuclease PH (EC 2.7.7.56) | |
| ATP-dependent HSL protease ATP-binding subunit HslU | |
| Heat shock protein 60 family chaperone GroEL | |
| Heat shock protein 60 family co-chaperone GroES | |
| Heat shock protein | |
| Oxidative stress | Catalase (EC 1.11.1.6) |
| Peroxidase (EC 1.11.1.7) | |
| NAD-dependent protein deacetylase of the SIR2 family | |
| Thioredoxin reductase (EC 1.8.1.9) | |
| Superoxide dismutase [Fe] (EC 1.15.1.1) | |
| Ferric uptake regulation protein FUR | |
| Nicotinamidase (EC 3.5.1.19) | |
| Superoxide dismutase [Cu-Zn] precursor (EC 1.15.1.1) | |
| NAD-dependent glyceraldehyde-3-phosphate dehydrogenase (EC 1.2.1.12) | |
| Zinc uptake regulation protein ZUR | |
| Carbon starvation | Carbon starvation protein A |
| Carbon storage regulator | |
| Osmotic stress | Sodium-dependent phosphate transporter |
| Potassium efflux system KefA protein | |
| Potassium voltage-gated channel subfamily KQT | |
| Na+/H+ antiporter (subunit A, B, C, D, E, F and G) | |
| NhaA, NhaD and Sodium-dependent phosphate transporters | |
| Glycine betaine transporter OpuD | |
| DNA repair | DNA-3-methyladenine glycosylase II (EC 3.2.2.21) |
| Formamidopyrimidine-DNA glycosylase (EC 3.2.2.23) | |
| A/G-specific adenine glycosylase (EC 3.2.2.-) | |
| Endonuclease III (EC 4.2.99.18) | |
| DNA ligase (EC 6.5.1.1) | |
| DNA mismatch repair protein MutS | |
| DNA mismatch repair protein MutL | |
| RecA protein | |
| Recombination protein RecR | |
| DNA polymerase III subunits gamma and tau (EC 2.7.7.7) | |
| DNA polymerase I (EC 2.7.7.7) | |
| Single-stranded-DNA-specific exonuclease RecJ (EC 3.1.-.-) | |
| Single-stranded DNA-binding protein | |
| Holliday junction DNA helicase RuvB | |
| Holliday junction DNA helicase RuvA |
Genome and potential genes in P. taiwanensis AL17
Phylogenomic analysis of the genome was carried out using the genomic sequences of the other Pseudoxanthomonas (Figure 2). Out of 129 species, only 28 were selected due to possessing the entire sequence. Furthermore, based on NCBI gene bank (National Center for Biotechnology Information) indicated only 7 species of Pseudoxanthomonas genus with assembly-level complete genomes. The complex process of genome assembly is susceptible to gaps in DNA sequences and genetic variations of assembly genome sequences.54 Seven complete genomes of the Pseudoxanthomonas were discovered and compared (Table 3). The data showed that the AL17 was the first species of P. taiwanensis to possess a complete genome sequence.
Table (3):
Genome data sets of AL17 compare to the other Pseudoxanthomonas genus
| Accession Number | Isolate | Source | Genome Size (Mb) | GC % | Amount | |||
|---|---|---|---|---|---|---|---|---|
| Gen | CDS | rRNA | tRNA | |||||
| – | AL17 | Compost | 3.1 | 72.1 | 2,833 | 2,775 | 6 | 58 |
| GCF_014397415.1 | P. mexicana GTZY2 | Wastewater | 4.0 | 67.0 | 3,724 | 3,664 | 6 | 50 |
| GCF_000233915.3 | P. spadix BD-a59 | Contaminated soil with BTEX (Benzene, Toluene, Ethylbenzene, Xylene) | 3.5 | 67.5 | 3,203 | 3,144 | 3 | 50 |
| GCF_022637495.1 | P. daejeonensis S9-A64R | Soil grassland | 3.5 | 68.5 | 3,160 | 3,108 | 3 | 45 |
| GCF_027941875.1 | P. mexicana DMF5 | Sediment | 3.9 | 67.0 | 3,717 | 3,656 | 6 | 51 |
| GCF_000972865.1 | P. suwonensis J1 | Soil | 3.9 | 70.0 | 3,322 | 3,268 | 3 | 47 |
| GCF_000185965.1 | P. suwonensis 11-1 | Compost | 3.4 | 70.0 | 3,167 | 3,105 | 6 | 52 |
| GCF_030322905.1 | P. winnipegensis ZSY-33 | Sediment | 4.5 | 69 | 3,905 | 3,842 | 6 | 53 |
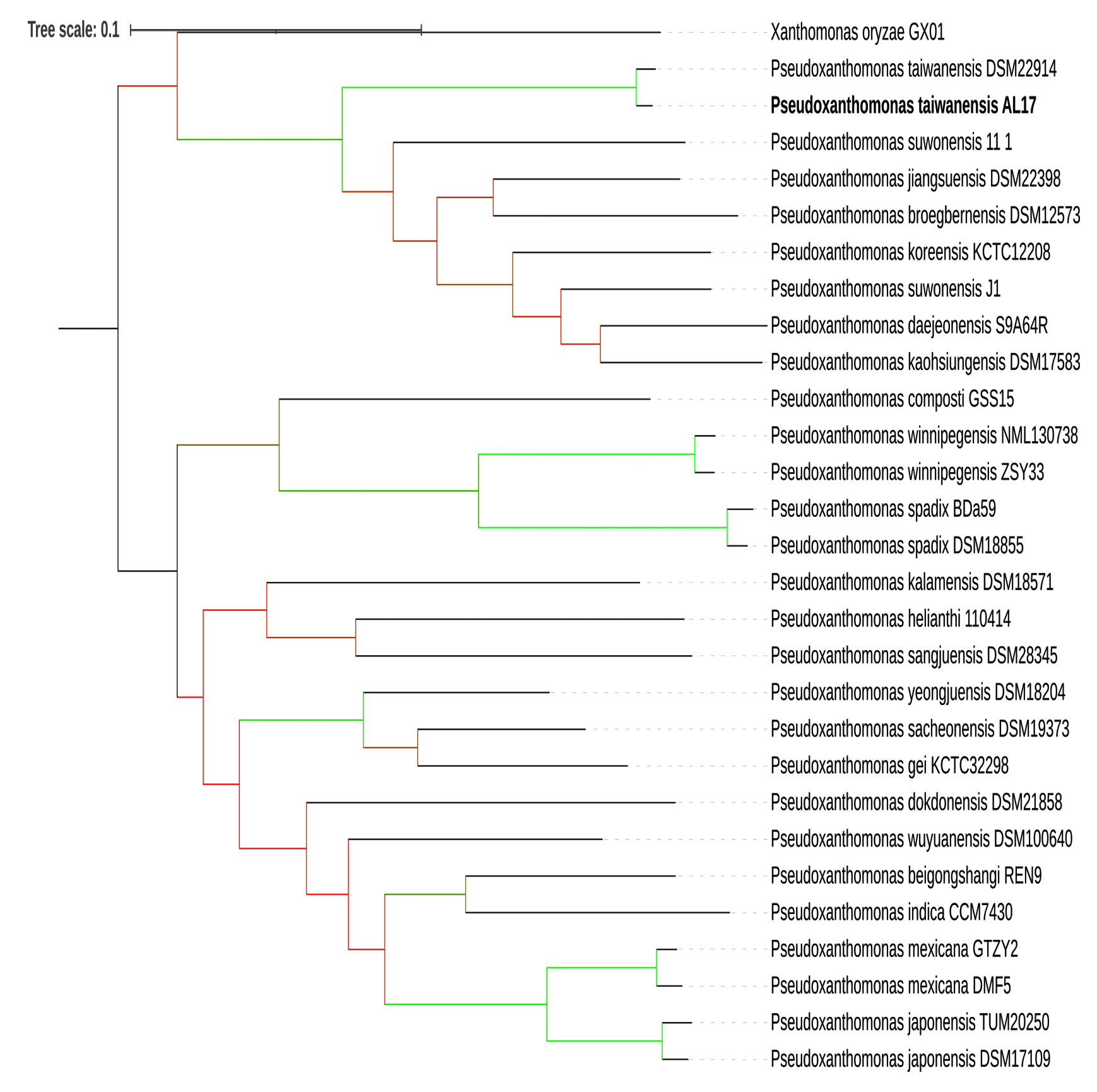
Figure 2. Phylogenomic analysis of the P. taiwanensis. Genome comparison to the other Pseudoxanthomonas genomes
Overall Genome Relatedness Index (OGRI) was used to compare 8 complete genome species. ANI and digital DNA-DNA hybridization (dDDH) programs confirmed that P. taiwanensis AL17 is significantly different from the other Pseudoxanthomonas genus (Figure 3). OGRI calculation provides valuable insights into the extent of phylogenomic relationships among the studied organisms.55 Cluster of Genes (COG) analysis, identified 391 genes from P. taiwanensis AL17 genome (Figure 4). 23.27% of these genes (91 out of 391) were not found in the COG category (Table 4), indicating the presence of novel genes in the organisms. Furthermore, 0.65% of the genes (7 out of 391) were categorized as genes S (signifying genes with unknown functions). Meanwhile, 15% of the genes are categorized as J gene, related function on translation, ribosome structure, and biogenesis. Several bacteria isolated from compost exhibited a high abundance of COG categories related to amino acid metabolism (E), energy production/conversion (C), transport of inorganic ions (P), carbohydrate transport and metabolism (G).31 Meanwhile, the P. taiwanensis AL17 genome showed Y category (nuclear structure), Z (cytoskeleton), W (extracellular structures), and B (chromatin structure and dynamics).
Table (4):
Number of genes annotated as COG in P. taiwanensis AL17
| General Function | COG Category | COG Description | Number of Gene | Percentage (%) |
|---|---|---|---|---|
| Cellular Process and Signaling | M | Cell Wall/Membrane Biogenesis | 75 | 7.00% |
| T | Signal Transduction Mechanism | 35 | 3.27% | |
| O | Post-translational Modification, Protein Turnover, Chaperones | 79 | 7.38% | |
| V | Defense Mechanisms | 28 | 2.61% | |
| U | Intracellular Exchange and Secretion, and Vesicular Transport | 18 | 1.68% | |
| D | Cell Cycle Control, Cell Division, and Chromosome Partitioning | 18 | 1.68% | |
| N | Cell Motility | 20 | 1.87% | |
| W | Extracellular Structure | 0 | 0 | |
| Y | Nuclear Structure | 0 | 0 | |
| Z | Cytoskeleton | 0 | 0 | |
| X | Mobilom: Prophages, transposons | 40 | 3.73% | |
| Storage and Information Processing | K | Transcription | 39 | 3.64% |
| L | Replication, Recombination, and Repair | 62 | 5.79% | |
| J | Translation, Ribosome Structure, and Biogenesis | 164 | 15.31% | |
| B | Chromatin Structure and Dynamics | 0 | 0 | |
| A | RNA Processing and Modification | 1 | 0.09% | |
| Metabolism | E | Amino Acid Transport and Metabolism | 93 | 8.68% |
| G | Carbohydrate Transport and Metabolism | 50 | 4.67% | |
| P | Inorganic Ion Transport and Metabolism | 56 | 5.23% | |
| C | Energy Production and Conversion | 79 | 7.38% | |
| H | Transport and Metabolism of Cofactors | 72 | 6.72% | |
| I | Transport and Metabolism of Lipids | 64 | 5.98% | |
| F | Transport and Metabolism of Nucleotides | 39 | 3.64% | |
| Q | Biosynthesis, Transport, and Catabolism of Secondary Metabolites | 8 | 0.75% | |
| Uncharacterized | S | Function Unknown | 7 | 0.65% |
| R | Predict General Function | 24 | 2.24% |
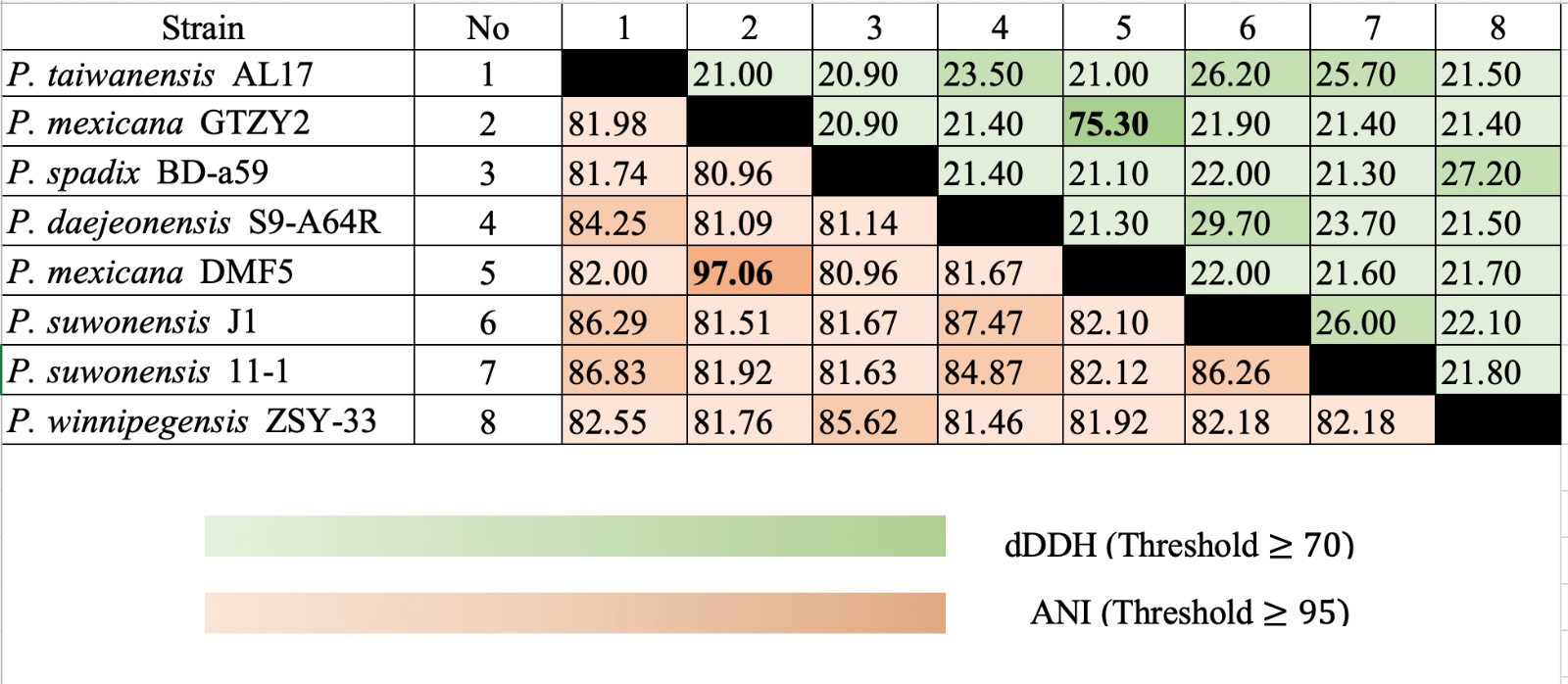
Figure 3. ANI and dDDH calculation of 7 Pseudoxanthomonas genomes for sample identification
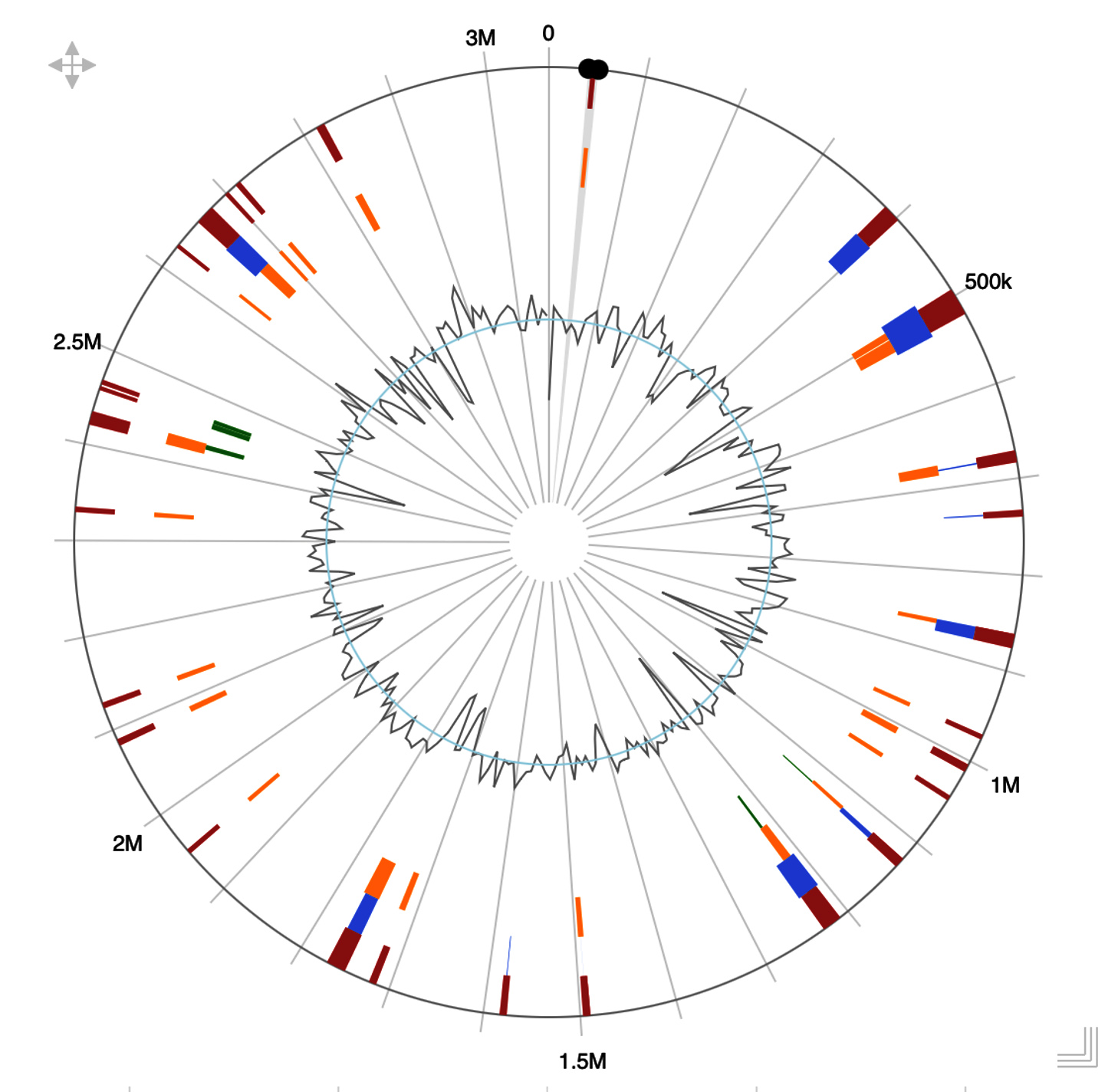
Figure 4. Genomic Island within P. taiwanensis AL17 using IslandViewer 4.0 version
Further analysis using the RAST program showed the presence of several potential enzymes (Figure 5). Among the enzymes were hydratase (EC 4.2.1.17) such as enoyl-CoA hydratase, dehydrogenase (EC 1.1.1.1) such as alcohol dehydrogenase and exopeptidases including aminopeptidase and D-alanyl-D-alanine carboxypeptidase (EC 3.4.16.4). The other are Hydrolases such as esterase, lipase, L-asparaginase, catalase, alpha-glucosidase, proteases, peroxidases, phospholipases A, C, and D, nucleases, transferases, and others were also found on the genome (Table 5). Furthermore, there are exciting discoveries on the presence of lipase genes. P. taiwanensis AL17 genome does not contain a true lipase gene, but rather a lipolytic enzyme within the GDSL type lipase/esterase motif. The GDSL type lipase/esterase is the lipolytic enzyme belonging to family II. The properties of the GDSL motif are characteristic of a novel lipase with several advantages compared to lipase with other motifs GXSXG.55 The GDSL motif is a unique subfamily of hydrolytic/lipolytic enzymes exhibiting different motifs of the GXSXG lipase with the serine active site near the N-terminal.55 Originally P. taiwanensis AL17 is from a mixture of household compost and dry leaves at the Waste Disposal Site in Saraga, ITB, Bandung.5 Therefore, the waste mixture had low fat content, leading to shorter thermogenic phases and indicating low chemical energy content of fats.6 Fats and oils were organic components dominant of waste in the composting process.56 Various chain lengths were found in the biodegradation of fats with saturated and unsaturated carbon chains.56-58 In addition, the GDSL enzyme shows a flexible active site that appears to change conformation in the presence and binding of different substrates, which agrees with the induced match mechanism proposed by Koshland.55
Table (5):
Potential genes on P. taiwanensis AL17 genome
| Feature ID | Genes |
|---|---|
| Hydratase | Aconitate hydratase 2 (EC 4.2.1.3) |
| Fumarate hydratase class II (EC 4.2.1.2) | |
| 2-methylisocitrate dehydratase (EC 4.2.1.99) | |
| 2-methylcitrate dehydratase FeS dependent (EC 4.2.1.79) | |
| Enoyl-CoA hydratase (EC 4.2.1.17) | |
| Pterin-4-alpha-carbinolamine dehydratase (EC 4.2.1.96) | |
| dTDP-glucose 4,6-dehydratase (EC 4.2.1.46) | |
| 3-hydroxyacyl-[acyl-carrier-protein] dehydratase, FabA form (EC 4.2.1.59) | |
| 3-hydroxydecanoyl-[ACP] dehydratase (EC 4.2.1.60) | |
| 3-dehydroquinate dehydratase II (EC 4.2.1.10) | |
| Urocanate hydratase (EC 4.2.1.49) | |
| phosphogluconate dehydratase (EC 4.2.1.12) | |
| phenate dehydratase (EC 4.2.1.51) | |
| methylthioribulose 1-phosphate dehydratase (EC 4.2.1.109) | |
| Phosphopyruvate hydratase (EC 4.2.1.11) | |
| Oleate hydratase (EC 4.2.1.53) | |
| Dihydroxy-acid dehydratase (EC 4.2.1.9) | |
| Threonine dehydratase (EC 4.3.1.9) | |
| Dehydrogenase | Malate dehydrogenase (EC 1.1.1.37) |
| L-threonine 3-dehydrogenase (EC 1.1.1.103) | |
| Aminomethyl-transferring glycine dehydrogenase (EC 1.4.4.2) | |
| Alanine dehydrogenase (EC 1.4.1.1) | |
| Isocitrate dehydrogenase (EC 1.1.1.41) | |
| Aspartate-semialdehyde dehydrogenase (EC 1.2.1.11) | |
| Glutamate-5-semialdehyde dehydrogenase (EC 1.2.1.41) | |
| UDP-N-acetylmuramate dehydrogenase (EC 1.3.1.98) | |
| Quinone-dependent dihydroorotate dehydrogenase (EC 1.3.5.2) | |
| 3-hydroxybutyrate dehydrogenase (EC 1.1.1.30) | |
| IMP dehydrogenase (EC 1.1.1.205) | |
| Dihydrolipoyl dehydrogenase (EC 1.8.1.4) | |
| Phosphoglycerate dehydrogenase (EC 1.1.1.95) | |
| Histidinol dehydrogenase (EC 1.1.1.23) | |
| 4-hydroxythreonine-4-phosphate dehydrogenase PdxA (EC 1.1.1.262) | |
| Choline dehydrogenase (EC 1.1.99.1) | |
| 3-hydroxyacyl-CoA dehydrogenase (EC 1.1.1.35) | |
| 3-isopropylmalate dehydrogenase (EC 1.1.1.85) | |
| dihydrolipoyl dehydrogenase (EC 1.8.1.4) | |
| glucose 1-dehydrogenase (EC 1.1.1.47) | |
| Exopeptidase | leucyl aminopeptidase (EC 3.4.11.1) |
| Prolyl aminopeptidase (EC 3.4.11.5) | |
| Endopeptidase La (EC 3.4.21.53) | |
| Type I methionyl aminopeptidase (EC 3.4.11.18) | |
| Xaa-Pro dipeptidase (EC 3.4.13.9) | |
| D-alanyl-D-alanine carboxypeptidase (EC 3.4.16.4) | |
| Peptidyl-dipeptidase Dcp (EC 3.4.15.5) | |
| Hydrolase Enzyme | Acyl-CoA thioesterase II (EC 3.1.2.-) |
| Arylesterase | |
| Carboxylesterase/lipase family protein | |
| Isoaspartyl peptidase/L-asparaginase | |
| Catalase (EC 1.11.1.6) | |
| amylo-alpha-1,6-glucosidase | |
| Carboxyl-terminal protease (EC 3.4.21.102) | |
| Peroxidase (EC 1.11.1.7) | |
| Thiol peroxidase, Bcp-type (EC 1.11.1.15) | |
| Phospholipase A1, C, D precursor (EC 3.1.1.32, EC 3.1.1.4) | |
| Acetyl-CoA acetyltransferase (EC 2.3.1.9) | |
| Dimethylallyltransferase (EC 2.5.1.1) | |
| glucans biosynthesis glucosyltransferase MdoH (EC 2.4.1.-) | |
| exodeoxyribonuclease VII small subunit (EC 3.1.11.6) | |
| Ribonuclease III (EC 3.1.26.3) | |
| Ribonuclease E (EC 3.1.26.12) | |
| GDSL-type esterase/lipase family protein |
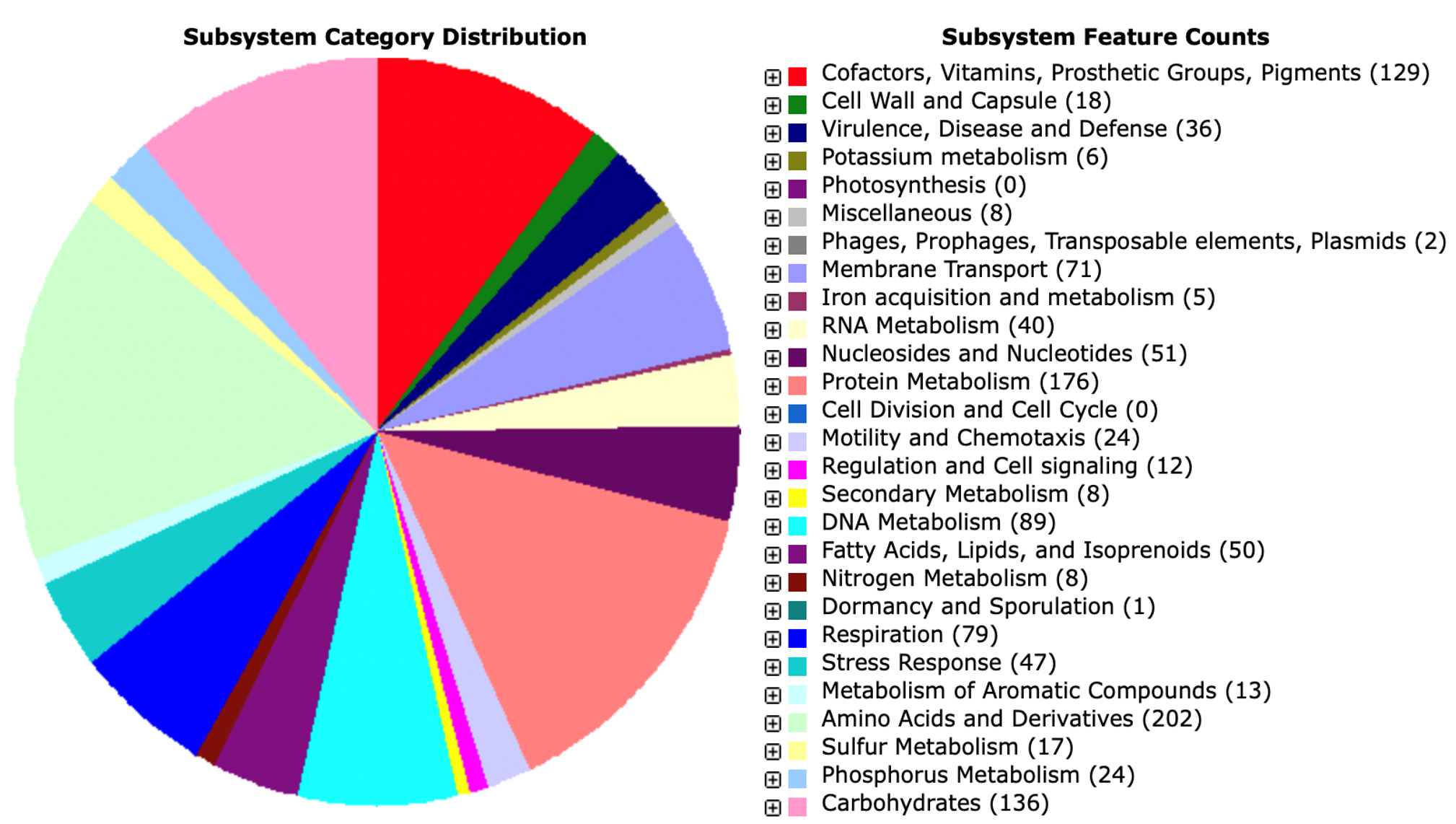
Figure 5. Overview of the subsystem categories genome P. taiwanensis AL17 using RAST server
The complete genome sequence of P. taiwanensis AL17 contains a genome length of 3,064,463 bp with a GC content of 72.08%. The genome comprises 2,833 CDSs, 6 RNA, 48 tRNA genes, and 17 pseudogenes. The genome reveals various potential genes for industrial application including hydratase, transferase, dehydrogenase, exopeptidase, and hydrolase. In addition, the genome exhibits various genes involved in stress-tolerant cell adaption. The information provides valuable insights to understand stress tolerant adaptation and to explore potential industrial application genes.
ACKNOWLEDGMENTS
The authors would like to acknowledge SIMLITABMAS “Doctoral Dissertation Research Program” Ministry of Education, Culture, Research and Technology, Republic of Indonesia.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
AUTHORS’ CONTRIBUTION
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
FUNDING
This work was supported by a grant from the SIMLITABMAS “Doctoral Dissertation Research Project” Ministry of Education, Culture, Research and Technology, Republic of Indonesia, contract No. 110/E5/PG.02.00.PL/2023.
AVAILABILITY OF DATA
All datasets generated or analyzed during this study are included in the manuscript.
ETHICS STATEMENT
This article does not contain any studies with human participants or animals performed by any authors.
- Edwards C. Isolation Properties and Potential Applications of Thermophilic Actinomycetes. Appl Biochem Biotechnol. 1993;42:161-179.
Crossref - Robinson CK, Webb K, Kaur A, et al. A major role for nonenzymatic antioxidant processes in the radioresistance of Halobacterium salinarum. J Bacteriol. 2011;193(7):1653-1662.
Crossref - Federici E, Pepi M, Esposito A, et al. Two-phase olive mill waste composting: Community dynamics and functional role of the resident microbiota. Bioresour Technol. 2011;102(23):10965-10972.
Crossref - Jurado M, Lopez MJ, Suarez-Estrella F, Vargas-Garcia MC, Lopez-Gonzalez JA, Moreno J. Exploiting composting biodiversity: Study of the persistent and biotechnologically relevant microorganisms from lignocellulose-based composting. Bioresour Technol. 2014;162:283-293.
Crossref - Syihab SF, Madayanti F, Akhmaloka. Isolation, Characterization and Identification of Lipolytic Thermophiles with Methanol Tolerance from Domestic Compost. J Pure Appl Microbiol, 2015;9(2):385-390.
- Chen MY, Tsay SS, Chen KY, Shi YC, Lin YT, Lin GH. Pseudoxanthomonas taiwanensis sp. nov., a novel thermophilic, N2O-producing species isolated from hot springs. Int J Syst Evol Microbiol. 2002;52(6):2155-2161.
Crossref - Norashirene MJ, Umi SH, Siti KMH, Nurdiana S. Biochemical Characterization and 16S rDNA Sequencing of Lipolytic Thermophiles from Selayang Hot Spring, Malaysia. IERI Procedia. 2013;5:258-264.
Crossref - Rawat N, Joshi GK. Bacterial community structure analysis of a hot spring soil by next generation sequencing of ribosomal RNA. Genomics. 2019;111(5):1053-1058.
Crossref - Bozan M, Akyol Ç, Ince O, Aydin S, Ince B. Application of next-generation sequencing methods for microbial monitoring of anaerobic digestion of lignocellulosic biomass. Appl Microbiol Biotechnol. 2017;101:6849-6864.
Crossref - Xu L, Wu YH, Zhou P, Cheng H, Liu Q, Xu XW. Investigation of the thermophilic mechanism in the genus Porphyrobacter by comparative genomic analysis. BMC Genomics. 2018;19(1):385.
Crossref - Palaniveloo K, Amran MA, Norhashim NA, et al. Food waste composting and microbial community structure profiling. Processes. MDPI AG. 2020;8(6):723.
Crossref - Biyada S, Merzouki M, Demcenko T, et al. Microbial community dynamics in the mesophilic and thermophilic phases of textile waste composting identified through next-generation sequencing. Sci Rep. 2021;11:23624.
Crossref - Geraldi A, Tay CC, Ni’matuzahroh, Fatimah, Hanafi WNW. Unraveling the bacterial diversity of cangar hot spring, indonesia by next generation sequencing of 16s rRNA gene. Biodiversitas. 2021;22(9):4060-4066.
Crossref - Saxena R, Chaudhary N, Dhakan DB, Sharma VK. Draft Genome Sequence of Gulbenkiania mobilis Strain MB1, a Sulfur-Metabolizing Thermophile Isolated from a Hot Spring in Central India. Genome Announc. 2015;3(6):e01295-15
Crossref - Bowman JL, Kohchi T, Yamato KT, et al. Insights into Land Plant Evolution Garnered from the Marchantia polymorpha Genome. Cell. 2017;171(2):287-304.e15.
Crossref - Saxena R, Dhakan DB, Mittal P, et al. Metagenomic analysis of hot springs in central india reveals hydrocarbon degrading thermophiles and pathways essential for survival in extreme environments. Front Microbiol. 2016;7.
Crossref - Wick RR, Judd LM, Holt KE. Performance of neural network basecalling tools for Oxford Nanopore sequencing. Genome Biol. 2019;20(1):129.
Crossref - De Coster W, D’Hert S, Schultz DT, Cruts M, Van Broeckhoven C. NanoPack: Visualizing and processing long-read sequencing data. Bioinformatics. 2018;34(15):2666-2669.
Crossref - Kolmogorov M, Yuan J, Lin Y, Pevzner PA. Assembly of long, error-prone reads using repeat graphs. Nat Biotechnol. 2019;37(5):540-546.
Crossref - Li H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34(18):3094-3100.
Crossref - Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25(7):1043-1055.
Crossref - Vaser R, Sovic I, Nagarajan N, Sikic M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 2017;27(5):737-746.
Crossref - Gurevich A, Saveliev V, Vyahhi N, Tesler G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics. 2013;29(8):1072-1075.
Crossref - Tatusova T, Dicuccio M, Badretdin A, et al. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016;44(14):6614-6624.
Crossref - Aziz RK, Bartels D, Best AA, et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genomics. 2008;9:75.
Crossref - Jain C, Rodriguez-R LM, Phillippy AM, Konstantinidis KT, Aluru S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat Commun. 2018;9(1):5114.
Crossref - Meier-Kolthoff JP, Carbasse JS, Peinado-Olarte RL, Goker M. TYGS and LPSN: A database tandem for fast and reliable genome-based classification and nomenclature of prokaryotes. Nucleic Acids Res. 2022;50(D1):D801-D807.
Crossref - Emms DM, Kelly S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019;20(1):238.
Crossref - Letunic I, Bork P. Interactive tree of life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021;49(W1):W293-W296.
Crossref - Bertelli C, Laird MR, Williams KP, et al. IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017;45(W1):W30-5.
Crossref - Antunes LP, Martins LF, Pereira RV, et al. Microbial community structure and dynamics in thermophilic composting viewed through metagenomics and metatranscriptomics. Sci Rep. 2016;6:38915.
Crossref - Tuomela M, Vikman M, Hatakka A, Itavaara M. Biodegradation of lignin in a compost environment: a review. Bioresource Technology. 2000;72(2):169-183
Crossref - Verma D, Kumar V, Satyanarayana T. Genomic attributes of thermophilic and hyperthermophilic bacteria and archaea. World J Microbiol Biotechnol. 2022;38(8):135.
Crossref - Brumm PJ, Gowda K, Robb FT, Mead DA. The complete genome sequence of hyperthermophile Dictyoglomus turgidum DSM 6724TM reveals a specialized carbohydrate fermentor. Front Microbiol. 2016;7:1979.
Crossref - Deckert G, Warren PV, Gaasterland T, et al. The complete genome of the hyperthermophilic bacterium Aquifex aeolicus. Nature. 1998;92:353-358.
Crossref - Sabath N, Ferrada E, Barve A, Wagner A. Growth temperature and genome size in bacteria are negatively correlated, suggesting genomic streamlining during thermal adaptation. Genome Biol Evol. 2013;5(5):966-977.
Crossref - Hickey DA, Singer GA. Genomic and proteomic adaptations to growth at high temperature. Genome Biology. 2004;5(10):117.
Crossref - Napoli A, Valenti A, Salerno V, et al. Functional interaction of reverse gyrase with single-strand binding protein of the archaeon Sulfolobus. Nucleic Acids Res. 2005;33(2):564-576.
Crossref - Bao Q, Tian Y, Li W, et al. A complete sequence of the T. tengcongensis genome. Genome Res. 2002;12(5):689-700.
Crossref - Armanet CB and Forterre P. Widespread distribution of archaeal reverse gyrase in thermophilic bacteria suggests a complex history of vertical inheritance and lateral gene transfers. Archaea. 2006;2(2):83-93.
Crossref - Nakashima M, Yamagami R, Tomikawa C, et al. Long and branched polyamines are required for maintenance of the ribosome, tRNAHis and tRNATyr in Thermus thermophilus cells at high temperatures. Genes to Cells. 2017;22(7):628-645.
Crossref - Peak MJ, Robb FT, Peak JG. Extreme Resistance to Thermally Induced DNA Backbone Breaks in the Hyperthermophilic Archaeon Pyrococcus furiosus. J Bacteriol. 1995;177.
Crossref - Grogan DW. Hyperthermophiles and the problem of DNA instability. Mol Microbiol. 1998;28(6):1043-1049.
Crossref - White MF. Archaeal DNA repair: paradigms and puzzles. Biochemical Society Transactions. 2003;31(3):690-693.
Crossref - Musgrave D, Forterre P, Slesarev A. Negative constrained DNA supercoiling in archaeal nucleosomes. Mol Microbiol. 2000;35(2):341-349.
Crossref - Kumari K, Sharma P, Tyagi K, Lal R. Pseudoxanthomonas indica sp. nov., isolated from a hexachlorocyclohexane dumpsite. Int J Syst Evol Microbiol. 2011;61(9):2107-2111.
Crossref - Goh KM, Gan HM, Chan KG, et al. Analysis of Anoxybacillus genomes from the aspects of lifestyle adaptations, prophage diversity, and carbohydrate metabolism. PLoS One. 2014;9(3):e90549.
Crossref - Goh KM, Chan KG, Lim SW, et al. Genome analysis of a new Rhodothermaceae strain isolated from a hot spring. Front Microbiol. 2016;7:1109.
Crossref - Takami H, Takaki Y, Chee GJ, et al. Thermoadaptation trait revealed by the genome sequence of thermophilic Geobacillus kaustophilus. Nucleic Acids Res. 2004;32(21):6292-6303.
Crossref - Wang J, Goh KM, Salem DR, Sani RK. Genome analysis of a thermophilic exopolysaccharide-producing bacterium – Geobacillus sp. WSUCF1. Sci Rep. 2019;9(1):1608.
Crossref - Yildiz SY, Radchenkova N, Arga KY, Kambourova M, Toksoy OE. Genomic analysis of Brevibacillus thermoruber 423 reveals its biotechnological and industrial potential. Appl Microbiol Biotechnol. 2015;99(5):2277-2289.
Crossref - Aldsworth TG, Sharman RL, Dodd CER. Bacterial suicide through stress. Cell Mol Life Sci. 1999;56.
Crossref - Vorob’eva LI. Stressors, Stress Reactions, and Survival of Bacteria: A Review. Appl Biochem Microbiol. 2004;40(3):217-224.
Crossref - Dsouza M, Taylor MW, Turner SJ, Aislabie J. Genomic and phenotypic insights into the ecology of Arthrobacter from Antarctic soils. BMC Genomics. 2015;16(1).
Crossref - Akoh CC, Lee GC, Liaw YC, Huang TH, Shaw JF. GDSL family of serine esterases/lipases. Progress in Lipid Research. 2004;43(6):534-552.
Crossref - de Souza C da CB, Amaral SNMB, Lima ESA, Lima J de O, do Carmo MGF, Garcia AC. Relation between changes in organic matter structure of poultry litter and heavy metals solubility during composting. J Environ Manage. 2019;247:291-298.
Crossref - Maina S, Kachrimanidou V, Koutinas A. A roadmap towards a circular and sustainable bioeconomy through waste valorization. Curr Opin Sustain Chem. 2017;8:18-23.
Crossref - Soobhany N. Insight into the recovery of nutrients from organic solid waste through biochemical conversion processes for fertilizer production: A review. J Clean Prod. 2019;241.
Crossref
© The Author(s) 2024. Open Access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License which permits unrestricted use, sharing, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.