


ISSN: 0973-7510
E-ISSN: 2581-690X
Proteins are essential for the proper functioning of cells. The techniques of cloning and protein production have facilitated the advancement of various fields and the creation of specific proteins for industrial and therapeutic uses. The bacterium Aeribacillus pallidus, which is able to survive in extreme conditions, is being studied with a view to identifying its robust enzymes. The objective of this study was to clone the protease gene from the A. pallidus P18 strain into the SUMO vector and produce recombinant protein in Escherichia coli BL21 for protein production. The protease enzyme gene from the A. pallidus P18 strain was isolated and amplified by using PCR. The PCR product was transferred into the SUMO expression vector and amplified in One Shot® Mach1TM-T1R bacteria, followed by colony PCR. Plasmid isolation was performed after positive colony selection. Gene integration was confirmed by cross-PCR using the gene forward, and vector reverse primers. For expression, the plasmid was transferred to E. coli BL21 cells. Two cultures were induced with different IPTG concentrations (0.5 mM and 1 mM) to optimize protein production. Bacterial cells were lysed, and SDS-PAGE analysis was conducted. Purification involved cell lysate preparation and purification using a ProbondTM column. SDS-PAGE and Coomassie Brilliant Blue G-250 staining confirmed successful purification. The results of this study indicate that the optimal product for protein production is that derived from a culture induced with 1 mM IPTG. Upon completion of the sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) procedure, the weight mass of the produced protein was determined to be 37 kDa, as indicated by the result of the gel stained with Coomassie brilliant blue G-250. This research successfully cloned the protease enzyme gene from the A. pallidus P18 strain using the pET-SUMO vector, performed purification and achieved the targeted result of protein production.
Protease Enzyme, Cloning, Protein Production, Aeribacillus pallidus P18
The production of recombinant proteins is one of the most important techniques used in molecular, medical, and industrial research.1 Recombinant DNA technology, which involves the incorporation of a modified vector into the host genome, alters the phenotype of the host.2 Therefore, the process requires the delivery of a foreign DNA fragment containing the desired gene into the host genome. Recombinant DNA technology is now widely used in almost every field of biological sciences.3
Enzymes play crucial roles in governing all biological processes in living organisms.4 In the past centuries, the number of industrial processes carried out by enzymes and the number of enzymes produced have increased.5 Currently, there is a renewed research effort worldwide to identify more sustainable and environmentally friendly biocatalytic processes. In this regard, remarkable studies are being conducted on the development of enzymes that can be produced in large quantities from biological sources and are resistant to high temperatures and various chemical environments in industrial applications using protein engineering and biotechnological methods.6,7 The most important microorganisms that produce alkaline protease enzymes are yeasts, fungi, and bacteria.8 Proteases are found in various forms and are produced by microorganisms, plants, and animals.9 Among them, bacterial proteases hold the most significant position due to their industrial applications.10,11 Due to their wide application in various industries such as food, detergent, leather, cheese, paper, pulp, and photographic films.9,12,13
Due to their ability to withstand harsh environments such as high temperatures, pressures, pH levels, and salt concentrations, thermophilic bacteria are crucial to industrial processes.14 The optimum growth temperature for the thermophilic bacterium Aeribacillus pallidus is between 55°C and 60°C. Higher processing temperatures can be used to increase reaction rates, increase the solubility of non-gaseous reactants and products, and reduce the possibility of microbiological contamination by mesophilic organisms. For these reasons, thermostable proteases are useful in some applications.15-17 In this study, the protease gene from the thermophilic bacterium A. pallidus P18 strain was cloned into a SUMO vector and produced the recombinant protein in the E. coli BL21 strain. Considering that most chemical reactions in industrial processes occur at high temperatures, it is anticipated that protease enzymes isolated from thermophilic bacteria could find easier application in such processes. In this article, we explored the utilization of the pET-SUMO system for recombinant protein expression. The potential of thermophilic bacterial protease enzymes in industrial processes will also be discussed, emphasizing their suitability for reactions conducted at elevated temperatures.
Bacterial strains, plasmids, medium, and reagents
Vectors were transformed using E. coli One Shot Mach1-T1R chemically competent cells, and E. coli BL21 (DE3) One Shot was utilized as the host strain for protein expression. T-A was cloned, and its protein was expressed using the pET SUMO vector from Invitrogen. Sigma, Fermantas, Merck, Gene Direx, and Promega provided all other compounds.
Isolation and molecular characterization of P18 from hot springs
Water samples taken from Pasinler (39° 58′ 37″ N 41° 39′ 56″ E) and Ilica (39° 56′ 17” N 41° 06′ 29″ E) hot springs situated in Erzurum province (Turkey) were brought to the laboratory under aseptic circumstances. The samples were serially diluted with sterile physiological water, and then they were spread on Tryptic Soy Agar mediums.18 The petri dishes were kept to incubation at 55°C for 24–48 h. Bacterial colonies growing on petri dishes were subcultured and purified. A total of 18 bacteria were purified, and the strain P18 displayed the highest protease activity. Then strain P18 was characterized. For this purpose, isolation of genomic DNA was performed according to the Promega WizardR Genomic DNA Purification Kit (A2360) protocol. Then, the 27F (5′-AGAGTTTGATCCTGGCTCAG-3′) and 1492R (5′-GGTTACCTTGTTACGACTT-3′) primers were used for 16S rRNA gene amplification.19 The amplified polymerase chain reaction fragments were cloned into E. coli JM101 strain with the pGEM-T Easy Cloning Vector (Promega, Southampton, UK) according to the manufacturer instructions.20,21 The sequences were compared with the other standard bacteria in EzTaxon, and the similarity rate was detected. Then, the GenBank accession number of the P18 was received. The phylogenetic tree was constructed using the Neighbor-Joining Technique with MEGA 4.22
Construction of expression plasmids
The coding sequence of the protease gene from the Aeribacillus pallidus P18 strain was cloned into the pET SUMO expression vector (Invitrogen) in order to create recombinant protein in prokaryote expression systems. Using the forward primer 5′-ATGTTTCACCGTACCGGGGAG-3′ and the reverse primer 5′-TCATGGCCGGACGACATGAAA-3′, the target gene’s coding sequence was amplified by PCR. PCR was performed at 95°C for 2 min, 35 cycles with denaturing at 94°C for 1 min, annealing at 58°C for 1 min, and extension at 72°C for 1.5 min, and the last cycle was at 72°C for 5 min. The PCR product was ligated into the vector pET SUMO.
Recombinant protein expression
Competent E. coli One Shot MachT1R cells were cultivated overnight at 37°C in 15 ml LB plates containing 50 mg/mL kanamycin. The cells had been treated with a ligation mixture. Colony PCR screening was used to find the DNA insert by examining the colonies. Using a ProbondTM purification Kit, plasmids were extracted from desired clones. Using the traditional heat shock technique, plasmids were converted into E. coli BL21 (DE3) and cultivated in LB plates. A single colony was inoculated into 15 mL of LB medium and cultured at 37°C and 200 rpm overnight to prepare the preculture. 2500 mL of the preculture were mixed to 100 mL of LB medium with 50 mg/mL kanamycin and 1% glucose in order to produce the 6xHis fusion protein. When the OD=600 value was roughly 0.4-0.6, 0.5 mM and 1 mM IPTG were added to the final concentration, and the medium was then incubated at 200 rpm and 37°C.23
Protein purification
Cells were collected after induction by centrifugation at 13000 rpm for one minute. Based on the previous studies, aiming to break down the cells as the pET-SUMO vector enables intracellular protein production, the following steps were performed. Firstly, 10 mL of lysis solution was added to the pellets, which were stored at -20°C. The samples were frozen for 5 seconds using liquid nitrogen and then allowed to thaw in a water bath set at 42°C after the sonication step. Afterward, the samples were centrifuged at maximum speed for 1 minute at 4°C. Filtrate from the cell homogenate was cleaned up using a ProBond Nickel-Chelating resin column. For purification our concentrated protein was incubated with Ni–chelating resin (2 mL) equilibrated in 10 mL denaturing binding buffer and allowed to bind to the Probond affinity column for 30 min at room temperature. All untagged proteins were allowed to pass through the column using pH 7.8 Guanidinium Lysis buffer (20 mM NaH2PO4, 6 M Guanidine Hydrochloride, and 500 mM NaCl), using pH 6 wash buffer (20 mM NaH2PO4 and 500 mM NaCl). The bound proteins were extracted from the column using pH 4 elution buffer (500 mM NaCl and 20 mM NaH2PO4). The final sample was used for SDS-PAGE analysis after being kept in ice-cold storage.
Protein analysis by SDS-PAGE
After the enzyme was purified, to determine the time-dependent expression level, purity, and molecular mass of the recombinant protein, a 3-8% batch SDS-PAGE was performed. The protein was stained with Coomassie Brilliant Blue G-250.
Effect of temperature on activity and stability of the purified protease
Temperature effects on the activity of the enzyme were measured at a range of temperatures, from 30°C to 90°C. The activity of the enzyme was assessed by letting the enzyme solution incubate at various temperatures (50-90°C) for 15, 30, 45, and 60 minutes in order to determine the temperature stability of the enzyme. The data obtained after the incubation was represented as the residual activity percentage (%) after the quantity of activity previous to the incubation at varied temperatures had been accepted as 100.
Molecular characterization of P18
The first stage of the present study focused on isolating thermophilic bacteria from Pasinler and Ilica hot springs, and a total of 18 bacteria were isolated. In the second stage, the protease production capabilities of these isolates were investigated. Strain P18 showed maximum protease activity. The P18 strain was identified as Aeribacillus pallidus according to the 16S rRNA gene sequence analysis. The phylogenetic tree was constructed using the Neighbor-Joining method (Figure 1).
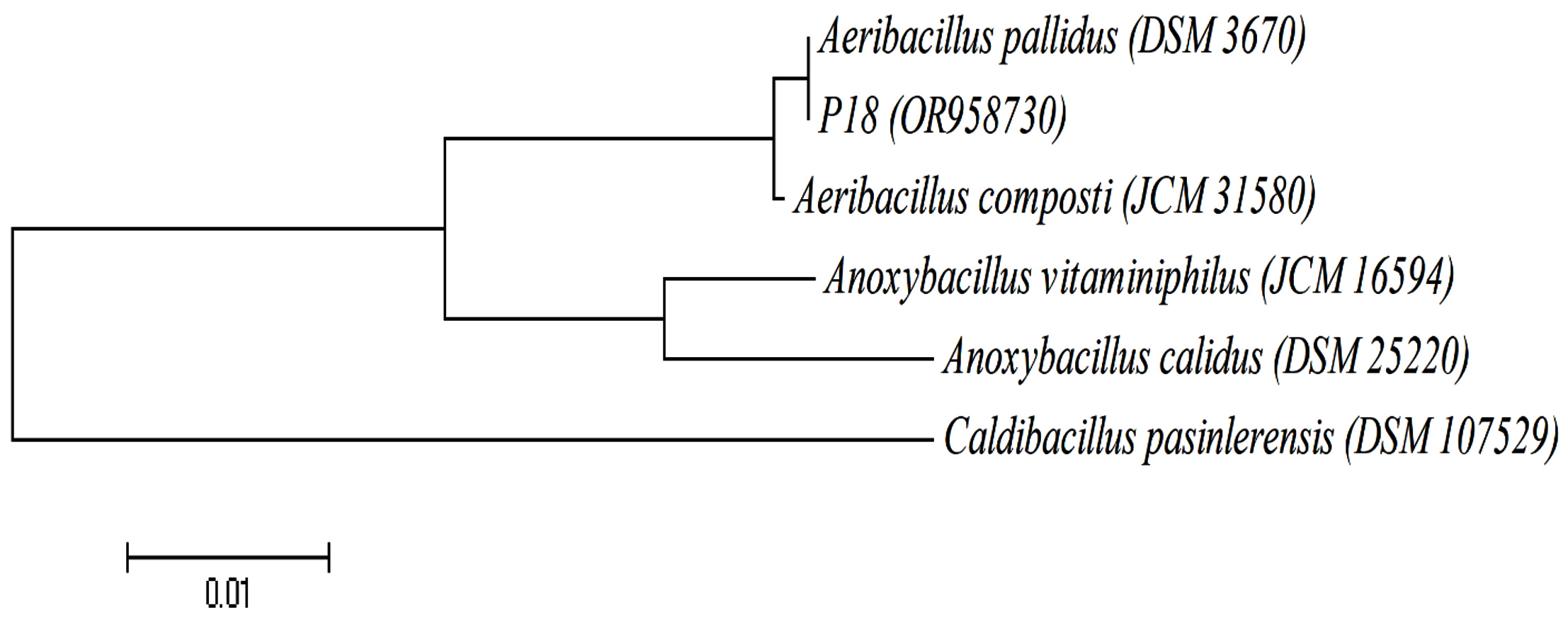
Figure 1. Phylogenetic relationships of P18 strain based on 16S rRNA gene sequence analyses. The tree was constructed by a neighbor-joining method. Bootstrap values were based on 100 replicates. Caldibacillus pasinlerensis was used as out-group. The scale bar represents 0.01 changes per nucleotide position
Construction of recombinant pET-SUMO-P18 vector
First, the serine protease gene region of the A. pallidus P18 strain was amplified, and it was detected the gene region was approximately 600 base pairs (Figure 2). A recombinant vector was created in accordance with the instructions. The protease gene from the P18 strain was ligated and cloned into the pET-SUMO vector following target protein gene sequence amplification and purification. The results of the colony PCR, cross PCR, and control PCR following transformation (Figures 3A, 3B, and 3C) demonstrated that the gene had been successfully introduced into the pETSUMO vector. As a result of sequence analysis, it was determined that the cloned region was 99% similar to Aeribacillus palidus (Figure 4).
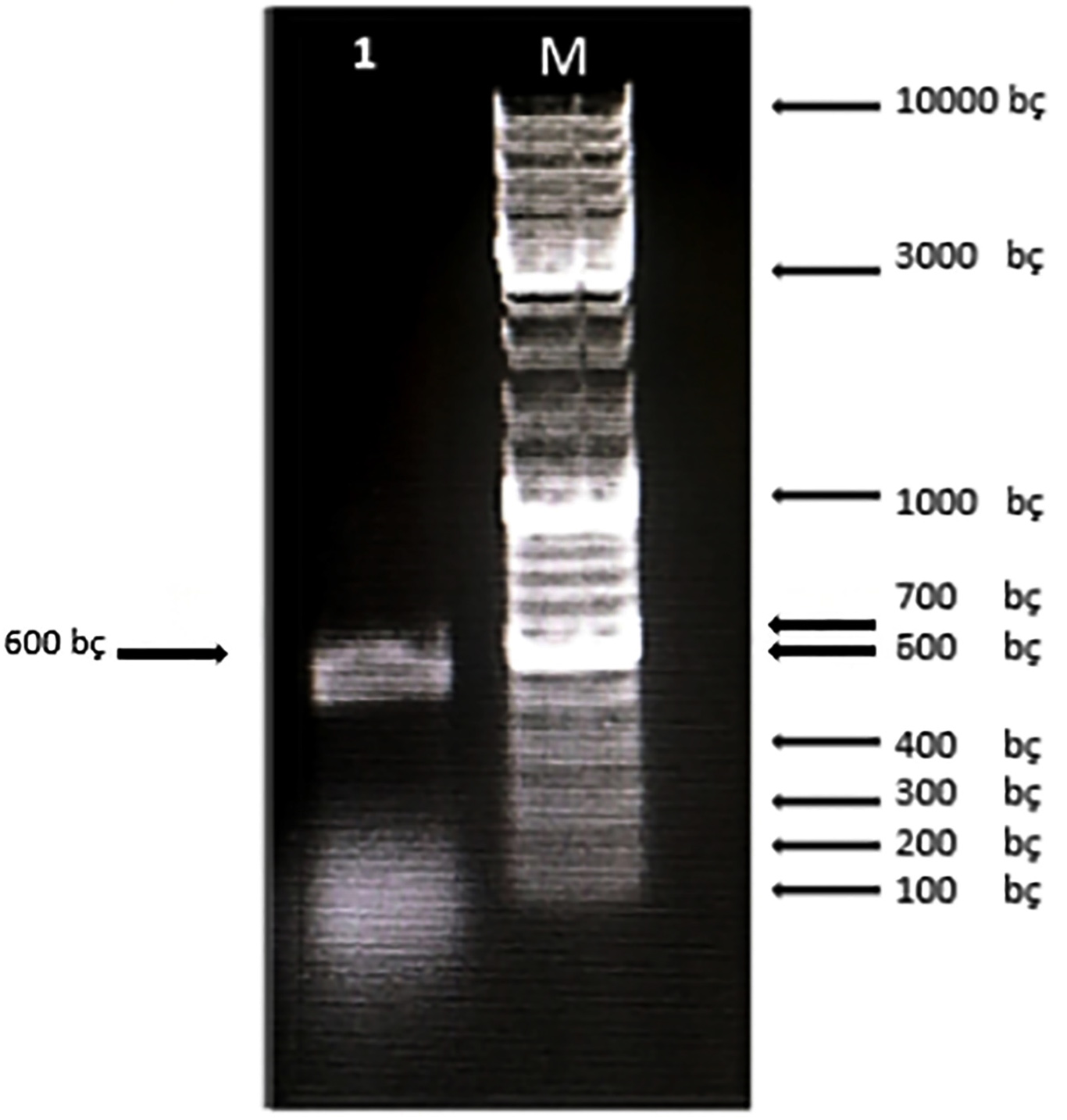
Figure 2. Electrophoresis of PCR products for P18 on 1% agarose gel electrophoresis
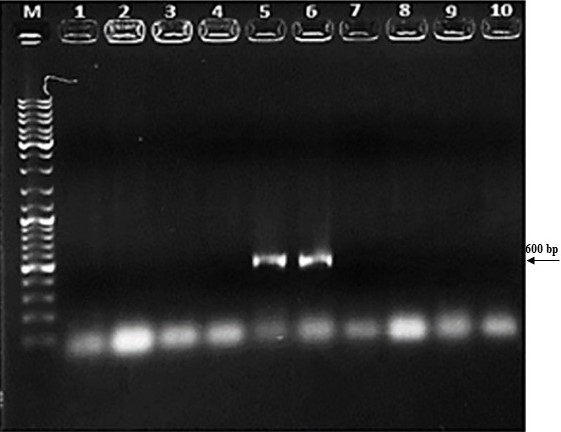
Figure 3A. Colony PCR analysis. M: Marker, Line 1-10: selected colonies
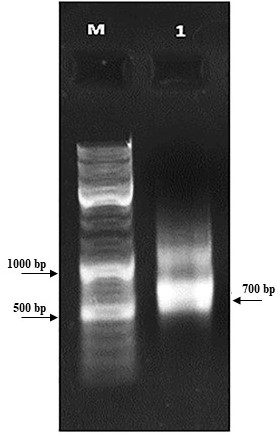
Figure 3B. Cross PCR analysis of purified recombinant pET-SUMO vector (M: Marker, 1: 5. gene forward and vector reverse primers)
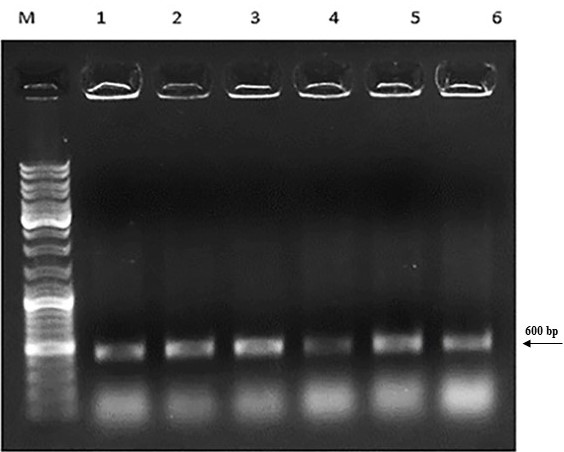
Figure 3C. Amplified products of P18 strain after transformation into BL21 bacteria (M: Marker, 1-6 lines: colony PCR products from BL21
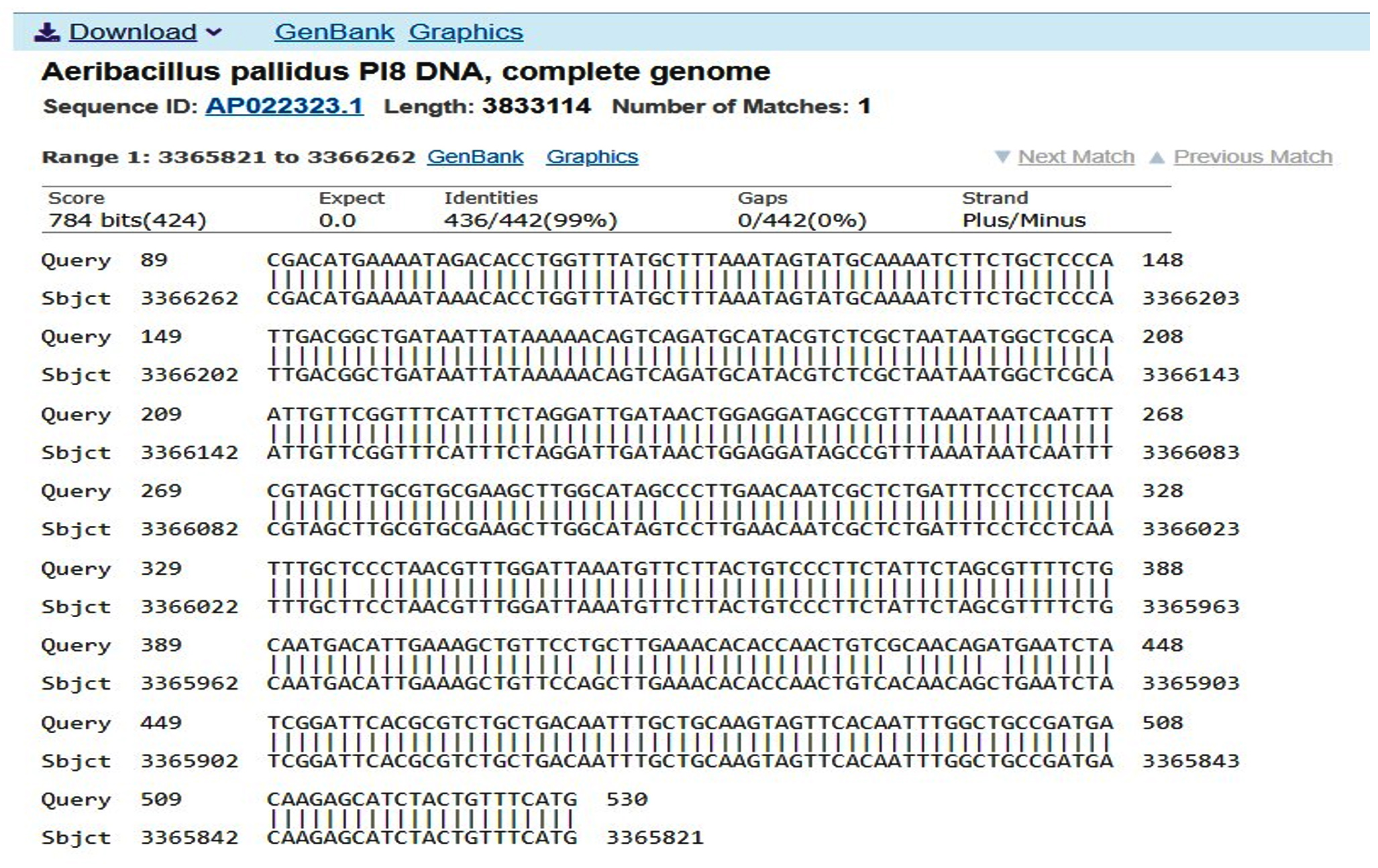
Figure 4. Sequence analysis results using forward and reverse primers to vector DNA
Expression of protease
In order to produce the protein, E. coli BL21 cells were transformed to express the coding areas of the A. pallidus P18 strain. IPTG was used to induce expression at final concentrations of 0.5 mM and 1 mM. Cells were extracted using a total of three cycles of fast freezing in liquid nitrogen and thawing in a 42°C water bath after IPTG induction. SDS-PAGE was used to evaluate the cell lysate. The outcomes demonstrate that the expression product’s molecular weight is 37 kDa. According to this finding, 37°C for 6 hours with 1 mM IPTG was chosen as the ideal expression condition.
Purification and identification of target protein
The cells were collected by centrifugation and lysed with a lysis solution under denaturing conditions. To break down the cells as the pET-SUMO vector enables intracellular protein production, 10 mL of lysis solution was added to the pellets, which were stored at -20°C. Following the sonication process, the samples were frozen in liquid nitrogen for 5 seconds before being allowed to defrost in a water bath set at 42°C. A sample was taken before purification, and SDS-PAGE was performed (Figure 5).
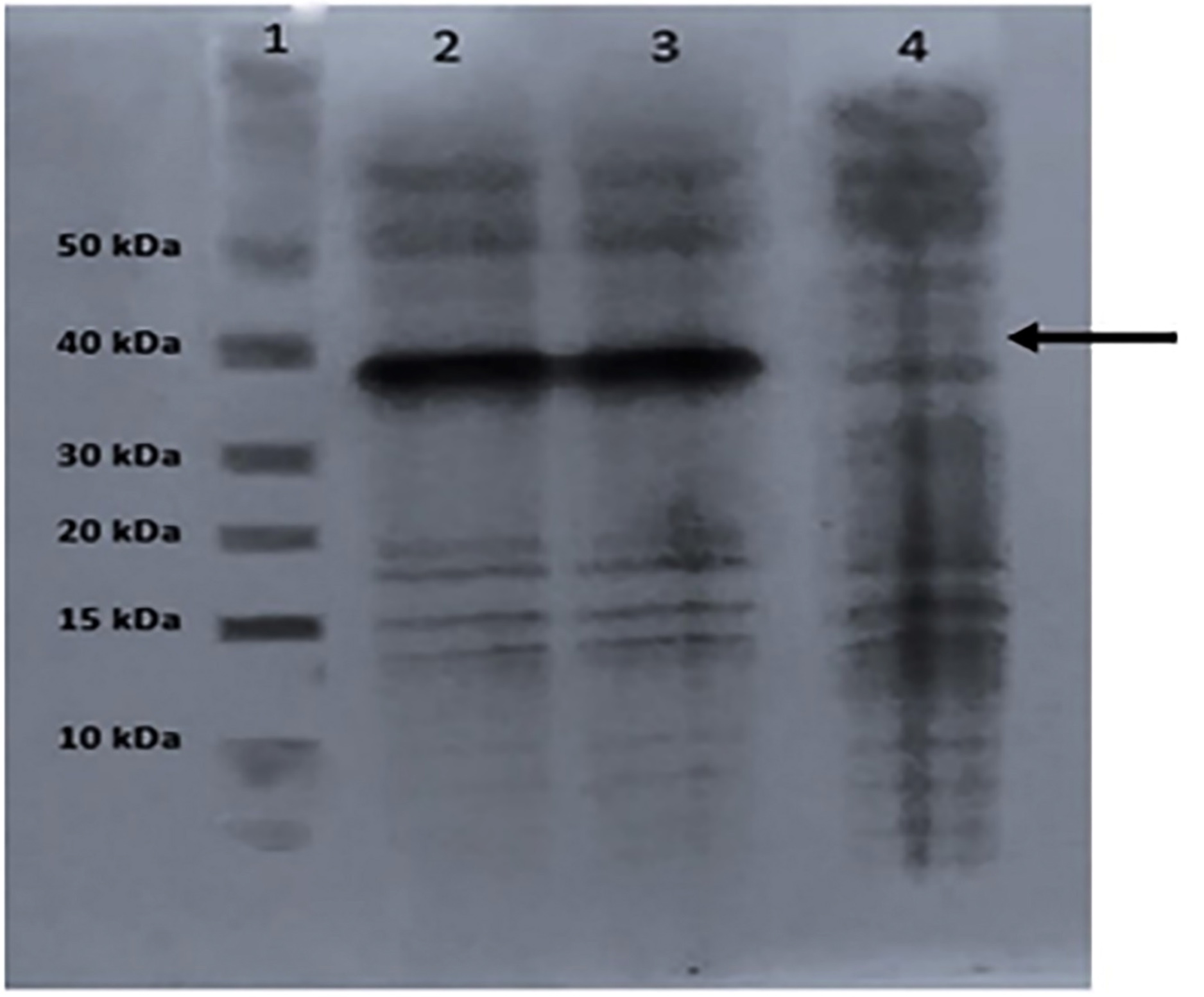
Figure 5. SDS-PAGE analysis of P18 pilot expression before purification (1: Marker, 2: Cell lysate induced with 0.5 mM IPTG, 3: Cell lysate induced with 1 mM IPTG, 4: Control (IPTG uninduced)
Then, the ProBond Nickel-Chelating resin kit was used to purify. Results from SDS-PAGE analysis show that the purified protein had a high expression level in the 1 mM IPTG-induced final expression (Figure 6), and the weight of the protein produced was 37 kDa based on the outcome of the gel stained with Coomassie brilliant blue G-250.

Figure 6. SDS-PAGE analysis of P18 pilot expression after purification (1: Marker, 2: Unpurified Cell Lysate Culture.3: Purified protein induced by 0.5 mM IPTG, 4: Purified protein induced by 1 mM IPTG)
Effect of temperature on activity and stability of the purified protease
Temperature plays an essential role in enzyme production by directly affecting the physiology and growth of microorganisms. As shown in Figure 7a, maximum enzyme activity was determined at 60°C. When the temperature stability profile was examined, it was determined that the enzyme had not lost more than 70% of its activity at 50-70°C temperatures. At 80 and 90°C, the enzyme lost more than 50% of its activity in 30 min (Figure 7b).
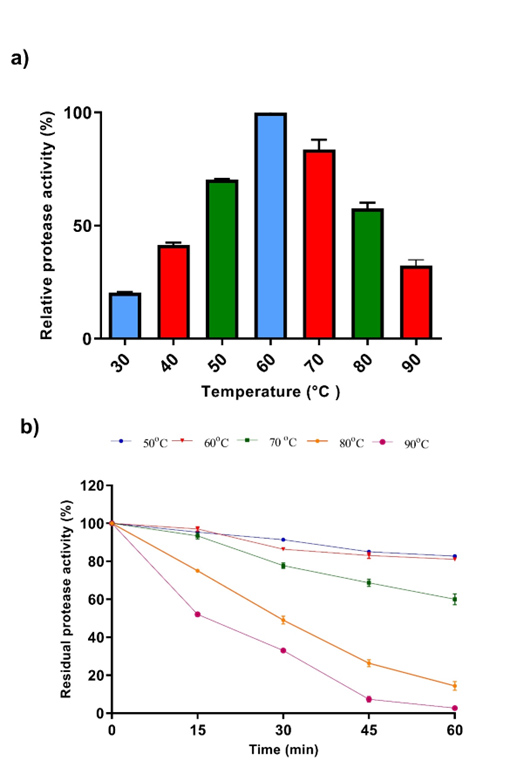
Figure 7. Effect of temperature on activity (a) and stability (b) of the purified protease from A. pallidus P18
Proteases have been successfully produced from many microbial sources, and about two-thirds of the commercial market consists of microbial proteases. Thermostability is an essential property for enzymes to achieve higher efficiency, protection from microbial contaminations, and fast conversion rates.24 Thermophilic microorganisms are the best sources of thermostable enzymes.25 Therefore, hot spring water was used for the isolation of thermostable protease-producing microorganisms. It was determined that the isolate P18 strain with the highest protease activity (P18) belonged to the Aeribacillus pallidus species.
When the literature data are examined, there are two studies on protease enzyme production and purification from A. palidus,26,27 while there is no study on protease cloning and expression from A. palidus.
Recombinant DNA technology is a technique widely used in molecular, medical, and industrial research.1 Forty years ago, Escherichia coli was the first host used for recombinant gene expression.28 Consequently, commonly reported expression systems are prokaryotic or mammalian cell expression systems.
Various methods and strategies are employed to produce recombinant proteins in our surroundings, aiming for time efficiency and low cost. The use of bacterial hosts is focused on obtaining large quantities of recombinant proteins rapidly and efficiently, with cost-effectiveness in mind.
Bacterial hosts possess invaluable characteristics for the rapid, efficient, and economical production of large quantities of recombinant proteins. These characteristics include rapid growth capability, the presence of diverse mutant strains, a well-known genome, and inexpensive requirements for optimal conditions.29 According to research, achieving the best-purified proteins has become a targeted goal in various sectors, such as the pharmaceutical and food industries.30 To improve these parameters, the use of expression partners in E. coli has emerged (also known as affinity tags or fusion tags).31
In this study, it was successfully cloned and produced serine protease from the A. pallidus P18 strain. In the literature, Through optimization of induction conditions, the culture induced with 1 mM IPTG demonstrated the highest protein yield. The estimated molecular mass of the produced protein was approximately 37 kDa. In the literature, many serine proteases with different molecular mass have been produced. For example, Kang et al. produced serine protease from the coryneform bacterium TU-19 with three different molecular weights (120 kDa, 80 kDa, and 45 kDa). In another study, the molecular weight of serine protease produced from Aeribacillus pallidus was 38 kDa.27
In this study, the gene responsible for the synthesis of the serine protease enzyme in Aeribacillus pallidus P18, isolated from a hot water source, was cloned and the related protein was purified. It was observed that the purified enzyme retained most of its activity up to 80°C and showed maximum activity at 60°C. These properties suggest that it is a suitable candidate for various applications where high temperature is required. However, further research is needed on the functional analysis and potential applications of the thermophilic protease.
ACKNOWLEDGMENTS
None.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
AUTHORS’ CONTRIBUTION
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
FUNDING
This research was supported by Ataturk University, project no. FYL-2021-9633.
DATA AVAILABILITY
All datasets generated or analyzed during this study are included in the manuscript.
ETHICS STATEMENT
This article does not contain any studies with human participants or animals performed by any of the authors.
- Sorensen HP. Towards universal systems for recombinant gene expression. Microb Cell Fact. 2010;9:27.
Crossref - Begna T and JC Okonkwo. Role of Recombinant DNA Technology in Agriculture. Int J Agri Biosci, 2020;9(5): 254-259.
- Sayed N, Allawadhi P, Khurana A, et al. Gene therapy: Comprehensive overview and therapeutic applications. Life sciences. 2022;294:120375.
Crossref - Gifre L, Aris A, Bach A, Garcia-Fruitos E. Trends in recombinant protein use in animal production. Microb Cell Fact. 2017;16(1):40.
Crossref - Patel AK, Dong C-D, Chen C-W, Pandey A, Singhania RR. Production, purification, and application of microbial enzymes. Biotechnology of Microbial Enzymes. 2023:25-57.
Crossref - Ismail AR, Kashtoh H, Baek K-H. Temperature-resistant and solvent-tolerant lipases as industrial biocatalysts: Biotechnological approaches and applications. Int J Biol Macromol. 2021;187:127-142.
Crossref - Victorino da Silva Amatto I, Gonsales da Rosa-Garzon N, Antonio de Oliveira Simoes F, et al. Enzyme engineering and its industrial applications. Biotechnol Appl Bioc. 2022;69(2):389-409.
Crossref - Matkawala F, Nighojkar S, Kumar A, Nighojkar A. Microbial alkaline serine proteases: Production, properties and applications. World J Microbiol Biotechnol. 2021;37(4):1-12.
Crossref - Naveed M, Nadeem F, Mehmood T, Bilal M, Anwar Z, Amjad F. Protease-a versatile and eco-friendly biocatalyst with multi-industrial applications: an updated review. Catalysis Letters. 2021;151(6):307-323.
Crossref - Haderer M, Neubert P, Rinner E, et al. Novel pathomechanism for spontaneous bacterial peritonitis: disruption of cell junctions by cellular and bacterial proteases. Gut. 2022;71(3):580-592.
Crossref - Yilmaz B, Baltaci MO, Sisecioglu M, Adiguzel A. Thermotolerant alkaline protease enzyme from A10: purification, characterization, effects of surfactants and organic solvents. J Enzym Inhib Med Ch. 2016;31(6):1241-1247.
Crossref - Wahab WAA, Ahmed SA. Response surface methodology for production, characterization and application of solvent, salt and alkali-tolerant alkaline protease from isolated fungal strain Aspergillus niger WA 2017. Int J Biol Macromol. 2018;115:447-458.
Crossref - Kirk O, Borchert TV, Fuglsang CC. Industrial enzyme applications. Curr Opin Biotech. 2002;13(4):345-351.
Crossref - Gupta GN, Srivastava S, Khare SK, Prakash V. Extremophiles: an overview of microorganism from extreme environment. Int J Agric Environ Biotechnol. 2014;7(2):371-380.
Crossref - Rahman RNZA, Razak CN, Ampon K, et al. Purification and Characterization of a Heat-Stable Alkaline Protease from Bacillus-Stearothermophilus F1. Appl Microbiol Biot. 1994;40(6):822-827.
Crossref - Singh J, Batra N, Sobti RC. Serine alkaline protease from a newly isolated Bacillus sp. SSR1. Process Biochem. 2001;36(8-9):781-785.
Crossref - Morabandza CJ, Dibangou V, Mabika FAS, et al. Optimization of Culture Conditions for Protease Production using Three Strains of Bacillus. J Pure Appl Microbiol. 2021;15(2):621-629.
Crossref - Baltaci MO, Ay H, Akbulut S, et al. Bacillus pasinlerensis sp. nov., a thermophilic bacterium isolated from a hot spring in Turkey. Int J Syst Evol Microbiol. 2020;70(6):3865-3871.
Crossref - Erkaya E, Genc B, Akbulut S, et al. Bacteriocin Producing Bacteria Isolated from Turkish Traditional Sausage Samples. J Pure Appl Microbio. 2020;14(2):1567-1576.
Crossref - Akbulut S, Baltaci MO, Adiguzel G, Adiguzel A. Identification and Potential Biotechnological Characterization of Lactic Acid Bacteria Isolated from White Cheese Samples. J Pure Appl Microbio. 2022;16(4):2912-2922.
Crossref - Albayrak S, Genc B, Ozkan H, Mesut T, Ahmet A. Presence of Different Bacterial Species in Thermal Sources and Novelty in Their Industrial Enzyme Productions. J Pure Appl Microbiol. 2019;13(3):1375-1387.
Crossref - Baltaci MO. Enhancement of cellulase production by co-culture of Streptomyces ambofaciens OZ2 and Cytobacillus oceanisediminis OZ5 isolated from rumen samples. Biocatal Biotransfor. 2022;40(2):144-152.
Crossref - Ceylan H, Erdogan O. Cloning, expression, and characterization of human brain acetylcholinesterase in using a SUMO fusion tag. Turk J Biol. 2017;41(1):77-87.
Crossref - Adamczak M, Krishna SH. Strategies for improving enzymes for efficient biocatalysis. Food Technol Biotech. 2004;42(4):251-264.
- Haki GD, Rakshit SK. Developments in industrially important thermostable enzymes: a review. Bioresource Technol. 2003;89(1):17-34.
Crossref - Mechri S, Berrouina MB, Benmrad MO, et al. Characterization of a novel protease from strain VP3 with potential biotechnological interest. Int J Biol Macromol. 2017;94(Part A):221-232.
Crossref - Yildirim V, Baltaci MO, Ozgencli I, Sisecioglu M, Adiguzel A, Adiguzel G. Purification and biochemical characterization of a novel thermostable serine alkaline protease from C10: a potential additive for detergents. J Enzym Inhib Med Ch. 2017;32(1):468-477.
Crossref - Cohen SN, Chang ACY, Boyer HW, Helling RB. Construction of Biologically Functional Bacterial Plasmids in-Vitro. P Natl Acad Sci USA. 1973;70(11):3240-3244.
Crossref - Swartz JR. Advances in Escherichia coli production of therapeutic proteins. Curr Opin Biotech. 2001;12(2):195-201.
Crossref - Demain AL, Vaishnav P. Production of recombinant proteins by microbes and higher organisms. Biotechnol Adv. 2009;27(3):297-306.
Crossref - Costa S, Almeida A, Castro A, Domingues L. Fusion tags for protein solubility, purification, and immunogenicity in : the novel Fh8 system. Front Microbiol. 2014;5:63.
Crossref
© The Author(s) 2024. Open Access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License which permits unrestricted use, sharing, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.