


ISSN: 0973-7510
E-ISSN: 2581-690X
Pre identified hydrocarbons degrading bacteria were used in this study, specific primer was conducted to amplification of AlkB gene, approximately 1206bp band size of this gene for Pseudomonas aeruginosa was detected and proofed by sequence and alignment analysis with NCBI database. The AlkB gene was inserted in PET-21a(+) plasmid vector as expression vector, then transformed in BL21(DE3) competent E. coli and confirmed by colony PCR technique using the T7 promoter and T7 terminator primers. The expression of the inserted gene was checked by determined the concentration of AlkB protein for multiple periods by Bradford assay method and the SDS-polyacrylamide gel electrophoresis method was revealed band of ̴46 KD molecular weight of the concerned protein. The gene amplification and cloning strategy was lay out before the practical part of the study by SnapGene software, this study was conducted to introduce cloned bacteria which facilitate the first step (key step) of alkane’s biodegradation and propose an appropriate strategy to construct genetically engineered microorganisms with multiple recombinant plasmid for enhance the degradation of the aliphatic fraction of hydrocarbon.
Cloning, AlkB gene, Pseudomonas aeruginosa.
Iraq is one of the main petroleum oil producers. As a result of this elevated production, the probability of accidents such as the spillage of oil and environmental pollution became obvious, particularly concerns regarding possible pollution of soil and subterranean water sources.
Hydrocarbons in different forms are one of the most important pollution sources of the environment, it is causing critical damages to the human being and the ecosystem, the most promising of these is bioremediation. The process of organisms usage or their enzyme systems to provide an effective alternative is called bioremediation1. The proportion of alkanes in crude oil is estimated about 20-50%. also, it is produced by living organisms as structural elements, waste, part of protection mechanisms or as a chemoattractants2. A lot of plasmids include genes which express of necessary enzymes for the important steps to bioremediation, for example enzymes essential in the biodegradation of naphthalene, toluene, octane etc. have been consider as plasmid encoded3. The techniques of molecular biology can be used to cut portions of DNA involves genes responsible for particular biodegradation pathways and insert it in plasmids. The recombinant plasmid can then be hosted into unique organisms to produce genetically engineered microorganisms (GEM) with new biodegradation abilities4.
The bacteria responsible for the degradation of the alkanes are widespread in the environment5. Bacteria genetically contained the Alk enzymes, which is involved in the in the metabolic steps of alkanes degradation, alkanes are the major compounds present in petroleum and its related components6. Alk enzyme system includes alkanes monooxygenases which is encoded by the AlkB genes, these genes are involved in the first hydroxylation step of the aerobic metabolism for aliphatic compounds, with the participation of the rubredoxin and rubredoxin reductase co-factors. The action of these enzymes is transport the electrons required in the AlkB function7, Some studies depend on AlkB gene as a bioindicator for the diversity of alkanes-degrading bacteria in the environment8 . In Pseudomonas, the responsible system for the first hydroxylation step of n-alkanes is known as alkane hydroxylase (monooxygenase) system9.
Molecular cloning allows combination of genes from different organisms for multiple purposes; the process depends on the following four main steps10:
1- Selection and isolation of a target gene to be cloned.
2- Selection the carrier (vector) of our target DNA sequence.
3- Ligation of the target to the vector followed by genetic transformation-vector set in to a host cell.
4- Selection host cell having the recombinant DNA.
The gene amplification and cloning strategy were lay out before the practical part of the study by SnapGene software 11 ,this study was conducted to facilitate the first step (key step) of alkanes biodegradation and propose an appropriate strategy to construct genetically engineered microorganisms with multiple recombinant plasmid for enhance the degradation of aliphatic fraction of hydrocarbon.
Hydrocarbons degrading bacteria and Molecular identification
In this study, pre-diagnosed hydrocarbons degrading bacterial strains were used, bacterial identification was done using 16S rRNA. The 16S rRNA genes were amplified by polymerase chain reaction (PCR) using a specific set of primer 27F 5’AGAGTTTGATCCTGGCTCAG’3 1492R 5’GGTTACCTTGTTACGACTT’312 and characterized by alignment with NCBI data bank13.
Amplification of AlkB gene
The polymerase chain reaction (PCR) was performed to amplify the AlkB of the isolated strains as shown in (Table 1) using the primes set supported with restriction sites of EcoRI in the forward primer plus (CAG) as flanking region and HindIII in the reverse primer plus (GAC) as flanking region, (Table 2) shows the PCR program details, the annealing temperature was fixed by gradient PCR.
Table (1):
Primers set for amplification of AlkB gene supported with restriction sites of EcoRI and (CAG) as flanking region in the 5 end of the forward primer and HindIII and (GAC) as flanking region in the 5 end of reverse one.
| AlkB gene primers | Sequence | GC content (%) | PCR product (bp) | Reference |
|---|---|---|---|---|
| Forward | 5’- cagGAATTCatgcttgagaaacacagagttc -3’ | 41% | 1218 | Current study |
| Reverse | 5’- gacAAGCTTctacgatgctaccgcagagg -3’ | 60% |
Construction protocol of the recombinant (PET-21a (+)-AlkB/BL21 (DE3) E. coli)
The amplification of alkB gene was performed using the primers supported with the RE sites of EcoRI (G↓AATTC) and HindIII (A↓AGCTT) (Bioneer, Korea) as shown in (Table 1). QIAGEN gel extraction Kit was used to purify the amplification product of PCR, and the PET-21a(+) (Novagen) plasmid was used as a vector. PCR products and plasmids were restricted with the same two endonucleases; EcoRI and Hind III at 37°C, and then T4 DNA ligase enzymes (Bioneer, Korea) were used in ligation process following the standard protocol from Bioneer company. In brief, the vector and the insert were mixed at a ratio of 1:5. The mixture was incubated at 16°C for 10min After heat inactivation at 65°C for 10 min, the reaction was chilled on ice14. The recombinant plasmid (PET-21a(+)-AlkB) was transformed to BL21(DE3) competent E. coli (Biolabs, New England) using heat shook method, briefly, 5μl of the ligation products and 50μl of competent DH5α cells were mixed and incubated for 30 min on ice, and subsequently heat shocked at 42°C for 10 sec and then placed back on ice. SOC medium (950μl) was added, and the transformed cells were incubated at 37°C for 60 min with agitation. After incubation, cells were pelleted and resuspended in 100µl SOC medium, which was then spread on LB agar plates containing ampicillin (100μg/ml). The plates were incubated overnight at 37°C 15, and then the PCR colony strategy was performed to recognise the recombinant bacterial cell using the T7 promoter and T7 terminator primers (Novagen) to amplify AlkB gene (Table 3).
Expression of the AlkB gene in PET-21a(+)-AlkB/BL21(DE3) E. coli
According to the modified procedure of the article17, IPTG (100µl/100ml) was used as an inducer and added when PET-21a(+)-AlkB/BL21(DE3) was at the logarithmic phase (OD600,0.4) in the LB broth medium with ampicillin(10µg ml-1). The experiment was carried out in three replicates [2 flasks (experiment and control) contains 100 ml for each replicate] using shaker incubator (250 rpm, 37˚C), the harvest process of the cells for each replicates were conducted at different times (2, 4 and 6 hours), the cell paste was pelleted by centrifugation (12000g, 10 min.) then kept at -20˚C till next experiment.
The precipitated cells were suspended in sterile ddH2O and boiled for 5min to disrupt the cells. Bradford assay method was used to determine the concentration of AlkB protein in the supernatant 18.
SDS-polyacrylamide gel electrophoresis
IPTG was used as inducer for different periods (4h, 6h, 8h) as well as the control (without inducer) was used too. About 120 V as operation voltage for 10% resolving gels and 80V applied to 5% stacking gels was used to perform SDS-polyacrylamide gel electrophoresis17,19. In each lane 15µl of protein was loaded. Coomassie Brilliant Blue G-250 was used to stain the gel.
Detection of the AlkB gene (amplification of the AlkB gene)
The oil degrading bacteria have been conducted to amplification using AlkB primer, the 1218 bp-band size for Alkb gene of Pseudomonas aeruginosa genomic DNA was detected on the agarose gel as shown in Fig. 1, there are two goals from the amplification process using these types of primers (table 2); the 1st goal was the detection of the gene while the 2nd one was to addition of EcoRI and Hind III restriction sites to the ends of gene for subsequent cloning protocol. Concerned gene sequence with terminal restriction sites was confirmed by the NCBI gene bank.
Table (2):
PCR amplification program of AlkB gene.
| stage | Temp. | time | cycles |
|---|---|---|---|
| pre-denaturation | 94°C | 5 min | 1 |
| denaturation | 94°C | 1 min | 35 |
| annealing | 50°C | 30 s | |
| extension | 72°C | 30 s | |
| final extension | 72°C | 7 min | 1 |
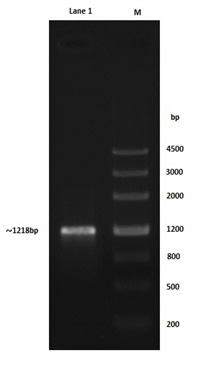
Fig. 1. Right lane; 4500bp ladder, left lane; ~1218 bp AlkB gene amplified with primers supported with EcoRI and HindIII restriction sites
Construction protocol of PET-21a(+)-AlkB plasmid vector
In this study, the 1206 bps AlkB gene was inserted into the MCS of the PET-21a(+) plasmid. Fig. 4 shows the recombinant plasmid map and complete protocol designed by SnapGene software which downloaded from the official website11. The pET plasmid vectors are the most effective system build up for the cloning and expression of recombinant proteins in E. coli. Target genes are inserted in pET plasmids and controlled by strong bacteriophage T7 transcription and (optionally) translation signals20. The name of the new recombinant plasmid vector was PET-21a(+)-AlkB.
Table (3):
T7 promoter and T7 terminator primers for PCR colony to detection of AlkB gene inserted in BL21 (DE3) competent E. coli.
| T7 primers set | Sequence | GC content (%) | PCR product (bp) | Reference |
|---|---|---|---|---|
| Forward T7 Promoter | 5-TAATACGACTCACTATAGGG-3 | 40% | 1452 | 16 |
| Reverse T7 Terminator | 5-GCTAGTTATTGCTCAGCGG-3 | 52% |
The recombinant plasmid vector PET-21a(+)-AlkB was transformed into competent E. coli BL21(DE3) ,the ampicillin(10µg/ml) was used to select the positive transformants because this plasmid have Ampicillin resistance gene (amp.R) which enable only E. coli harboring PET-21a(+) to grow in the medium.
Colony PCR technique was used to confirm the positive transformants colony harboring the PET-21a(+)-AlkB vectors, moreover the forward T7 promoter primer and reverse T7 terminator primer was used in PCR procedure to confirm that coding region of this gene is also under the transcriptional and translational control of the TEF1 promoter and terminator. The amplified segment was about 1452bp which is equal to that of the predicated fragment as shown in Fig. 2, the PET-21a(+) plasmid includes AlkB gene was called PET-21a(+)-AlkB vectors.
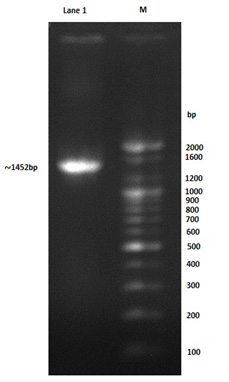
Fig. 2. Gel electrophoresis of colony PCR product using forward T7 promoter primer and reverse T7 terminator primer: 100bp marker, lane1: 1452bp amplified fragment
The band size (1452bp) and the analysis of the amplified gene sequencing result by using NCBI BLAST database proved the colony to be the AlkB cloned recombinant E. coli, the constructed bacteria was called PET-21a(+)-AlkB/BL21(DE3) E. coli.
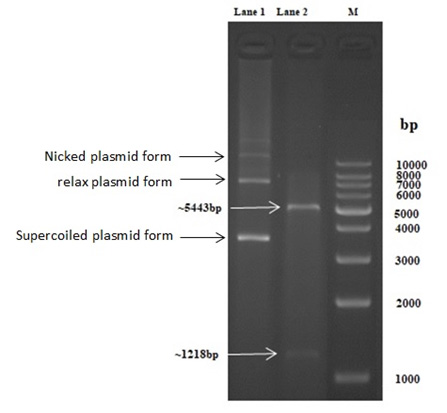
Fig. 3. Lane 1: Native PET-21a(+) plasmid, lane 2: linear PET-21a(+)-AlkB digested with EcoRI/HindIII, lane 3:1kb DNA ladder
Fig. 3 shows two bands in the lane 2 as a result for digestion of the PET-21a(+)-AlkB with EcoRI/HindIII restriction enzymes ,the lower band was equal to the AlkB gene size and the upper band was equal to the PET-21a(+) size while the lane 1 show about four bands as a result of the gel electrophoresis of the native uncut PET-21a(+) plasmid ,these bands related to supercoiled, relaxed, nicked respectively and that agree with21.
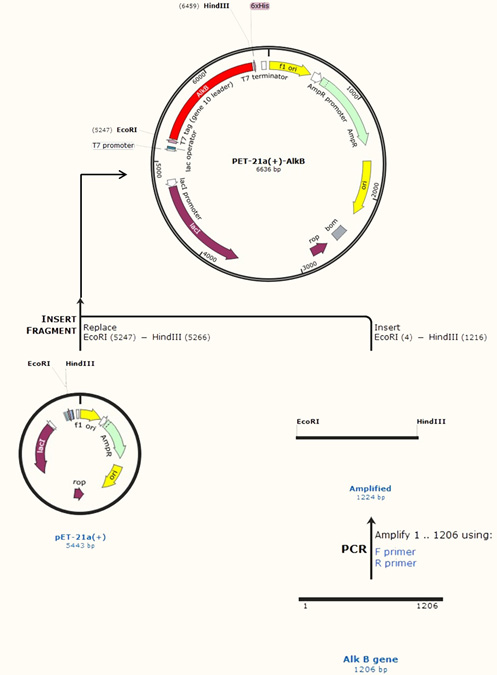
Fig. 4. Construction protocol of PET-21a(+)-AlkB plasmid vector
Expression of AlkB gene in PET-21a(+)-AlkB/BL21(DE3) E. coli
Bradford assay demonstrate that the crude protein was produced in each period of experiment (2h,4h,6h,) using IPTG as inducer (Fig. 5). protein concentration increased gradually with the time, but highest concentrations were detected at induction, so that there are significant differences in total protein production between the induce PET-21a(+)-AlkB/BL21(DE3) E. coli and the non-induced bacteria due to the high levels of AlkB protein produced as a result for IPTG induction which then confirmed by SDS page.
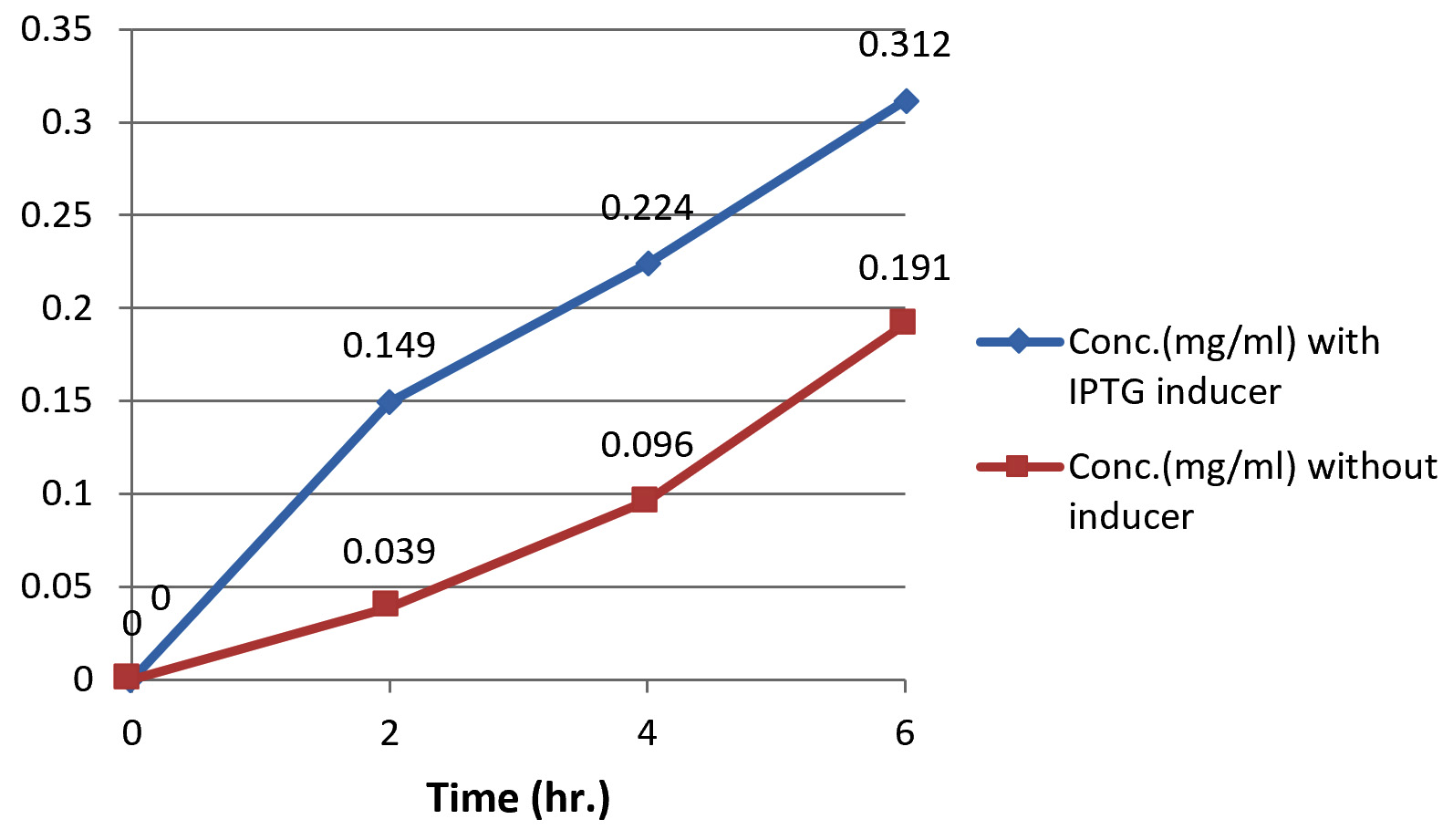
Fig. 5. Protein conc. at (2,4,6) hr. using IPTG as inducer
SDS-polyacrylamide gel electrophoresis
Approximately 46-48 KD of AlkB protein bands on the polyacrylamide gel were appear in all the times of the experiment using IPTG as inducer, the band size was closely related to the calculated value from the sequence of amino acids and agreed with22. The results shows, there is no significant differences between the different induction periods of IPTG (Fig. 6), which indicates that the minimum time of induction applied in the study was sufficient for bacteria to induce and to produce the recombinant protein, as well as the continuation of protein production with induction time indicates that the protein produced was not have toxic affection to the recombinant cells.

Fig. 6. Protein gel of AlkB with different induction times of IPTG. M: protein marker, 1: IPTG(4h),2: IPTG(6h), 3:IPTG(8h), 4:without inducer.
In conclusion, using of competent E. coli (BL21(DE3) and PET21a(+) system for expression of alkane hydroxylase-1 (ALKB1) suggestion a feasible and appropriate strategy for a wide conception of the catalytic mechanism of this interesting non-heme oxygenases. The newly synthesized alcohols don’t convert to subsequent aldehyde and fatty acid because the absence of alcohol oxidation enzymes in E. coli, therefore the downstream enzymes of the native strains is necessary to the function of recombinant alkB1.
The availability of different hydroxylases and catalyst enzymes (rubredoxin and rubredoxin reductase) will lead to enhancement of decontamination systems for more friendly environment studies which focus on bioremediation field. Moreover, there are many applications of biotransformation of common starting materials into fine chemical production related to this enzyme system.
ACKNOWLEDGMENTS
All thanks and appreciation to the staff of Marine bacteriology and agriculture biotechnology laboratories, which provided great support during the period of research.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
AUTHORS’ CONTRIBUTION
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
FUNDING
None.
ETHICS STATEMENT
Not applicable.
AVAILABILITY OF DATA
All datasets generated or analyzed during this study are included in the manuscript.
- Olukunle, O, Boboye, B. Phylogenetic analysis of Oil–degrading Bacteria Associated with Polluted Sites in River State Nigeria. Archives of Applied Science Research, 2012; 4(4): 1600-1608.
- van Beilen JB, Duetz, WA, Schmid A, Witholt B. Practical issues in the application of oxygenases. Trends in Biotechnology, 2003; 21(4): 170-177.
Crossref - (a) Gibson, DT. Microbial Degradation of Organic Compounds. Marcel Dekker Incorporated: 1984; (b) Neilson AH. The biodegradation of halogenated organic compounds. Journal of Applied Bacteriology, 1990; 69(4): 445-470.
Crossref - (a) Lee SG, Yoon BD, Park YH, Oh HM. Isolation of a novel pentachlorophenol-degrading bacterium, Pseudomonas sp. Bu34. J. Appl. Microbiol., 1998; 85(1): 1-8; Crossref (b) Akpe AR, Ekundayo AO, Esumeh FI, Degradation of crude oil bacteria: a role for plasmid borne genes. Global J. Sci. Front. Res., 2013; 13: 20-6.
- (a) Wentzel A, Ellingsen TE, Kotlar H-K, Zotchev SB, Throne-Holst M. Bacterial metabolism of long-chain n-alkanes. Applied Microbiology and Biotechnology, 2007; 76(6): 1209-1221; Crossref (b) Atlas RM, Aislabie J, Bej AK. Polar microbiology: the ecology, biodiversity and bioremediation potential of microorganisms in extremely cold environments. CRC Press: 2009.
- Rojo F. Degradation of alkanes by bacteria. Environmental Microbiology, 2009; 11(10): 2477-2490.
Crossref - Van Beilen JB, Funhoff EG, Alkane hydroxylases involved in microbial alkane degradation. Applied Microbiology and Biotechnology, 2007; 74(1): 13-21.
Crossref - (a) Kuhn E, Bellicanta GS, Pellizari VH. New alk genes detected in Antarctic marine sediments. Environmental Microbiology, 2009; 11(3): 669-673; (b) Perez-de-Mora A, Engel M, Schloter M. Abundance and diversity of n-alkane-degrading bacteria in a forest soil co-contaminated with hydrocarbons and metals: a molecular study on alkB homologous genes. Microbial Ecology, 2011; 62(4): 959-972.
Crossref - Van Beilen JB, Wubbolts MG, Witholt B. Genetics of alkane oxidation byPseudomonas oleovorans. Biodegradation, 1994; 5(3-4): 161-174.
Crossref - Rodrigues TB. Silva AET, Molecular Diversity of Environmental Prokaryotes. CRC Press: 2016.
Crossref - https://www.snapgene.com.
- Miyoshi T, Iwatsuki T, Naganuma T. Phylogenetic characterization of 16S rRNA gene clones from deep-groundwater microorganisms that pass through 0.2-micrometer-pore-size filters. Appl. Environ. Microbiol., 2005; 71(2): 1084-1088.
Crossref - http://www.ncbi.nlm.nih.
- Zeng F, Zang J, Zhang S, Hao Z, Dong J, Lin Y. AFEAP cloning: a precise and efficient method for large DNA sequence assembly. BMC Biotechnology, 2017; 17(1): 81.
Crossref - Zeng F, Zhang S, Hao Z, Duan S, Meng Y, Li P, Dong J, Lin Y. Efficient strategy for introducing large and multiple changes in plasmid DNA. Scientific Reports, 2018; 8(1): 1714.
Crossref - (a) Khani M-h, Bagheri M, Dehghanian A, Zahmatkesh A, Bidhendi SM, Najafabadi Z.S, Banihashemi R. Effect of C-Terminus Modification in Salmonella typhimurium FliC on Protein Purification Efficacy and Bioactivity. Molecular Biotechnology, 2019; 61(1): 12-19; Crossref (b) Brown A. Sequencing, sub-cloning, expression and purification of 2-hydroxychromene-2-carboxylate isomerase from Sphingomonas paucimobilis EPA505. 2008; 26.
- Luo Q, He Y, Hou D-Y, Zhang J-G, Shen X-R. GPo1 alkB gene expression for improvement of the degradation of diesel oil by a bacterial consortium. Brazilian Journal of Microbiology, 2015; 46(3): 649-657.
Crossref - (a) Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochemistry 1976; 72(1-2), 248-254; Crossref (b) Stoscheck C. Quantitation of Protein Methods in Enzymology. Pierce, 1990; 182: 50-69. Crossref
- Manns JM. SDS-Polyacrylamide Gel Electrophoresis (SDS-PAGE) of Proteins. Current Protocols in Microbiology, 2011; 22(1): A.3M.1-A.3M.13.
Crossref - Mierendorf RC. Morris BB, Hammer B, Novy RE. Expression and purification of recombinant proteins using the pET system. In The nucleic acid protocols handbook, Springer: 2000; 947-977.
Crossref - Magdeldin S. Gel electrophoresis: Principles and basics. BoD–Books on Demand: 2012.
Crossref - Xie M, Alonso H, Roujeinikova A. An improved procedure for the purification of catalytically active alkane hydroxylase from Pseudomonas putida GPo1. Applied Biochemistry and Biotechnology, 2011; 165: (3-4): 823-831.
Crossref
© The Author(s) 2020. Open Access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License which permits unrestricted use, sharing, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.