


ISSN: 0973-7510
E-ISSN: 2581-690X
This study examined culturable bacterial communities and physicochemical properties across ten governorates in the Makkah Region of Saudi Arabia, focusing on how environmental gradients shape bacterial community structure. A total of fifty soil samples were collected from coastal, peri-coastal, and inland locations. Standard protocols were used to measure soil pH, moisture content, and particle size distribution. Through serial dilution and inoculation on selective media, one hundred bacterial isolates were obtained. Phylogenetic relationships were inferred using 16S rRNA gene sequencing and neighbor-joining methods, and taxonomic assignments were further supported by secondary structure analysis. Soil pH ranged from 7.00-7.81, while moisture content differed by a factor of 24 (0.5%-12.0%) between governorates. The number of isolates abundance per governorate varied from 6 to 20, including three strains of Staphylococcus hominis and four Gram-negative taxa: Pseudomonas aeruginosa, Erwinia phyllosphaerae, Xanthomonas maliensis, and Dyella marensis. SNP analysis identified 3-14 nucleotide substitutions per isolate, corresponding to divergence levels of 0.29%-1.40%. Despite its low moisture content, Fair Capital Governorate yielded an unexpectedly large number of isolates, indicating possible human-driven alterations to soil conditions. SEM observations revealed marked differences in biofilm colonization patterns that correlated with soil texture and moisture levels. By integrating cultivation-based isolation, molecular identification, phylogenetic reconstruction, and secondary structure validation, this study advances understanding of bacterial diversity in the arid terrestrial habitats of Western Saudi Arabia and sheds light on microbial strategies for adapting to extreme environments.
Soil Microbiota, Bacterial Diversity, 16S rRNA Gene Sequencing, Makkah Region, Soil Physicochemical Properties
Soil microbiota are among the most numerous and diverse biotic communities on earth, with a number estimated to be about 10 billion microbial cells in one gram of soil.1 These microbiota are responsible for a number of critical ecosystem processes, including the recycling of nutrients, decomposition of organic matter, formation of soil, and promotion of healthy and productive plant growth.2 Thus, it is essential to understand the composition, diversity, and dynamics of soil microbiota to evaluate soil quality, predict soil responses to environmental stresses, and manage soil health in both agroecosystems and natural ecosystems.3,4 Desert and arid ecosystems present extreme environmental conditions, which significantly limit the diversity and functional complexity of microbiota.5 The defining features of these extreme environments include very low hydrologic availability, extremely hot temperatures, high rates of radiation, very low amounts of organic carbon (usually less than 0.1%), very high salt concentrations, and very low pH values.6,7 The extreme environmental conditions acting upon these ecosystems function as selective pressures, resulting in the development of specialized and highly tolerant microbial populations that can thrive under polyextremotolerant stress conditions.8 As such, the Kingdom of Saudi Arabia provides an excellent model system to study the adaptations of microbial populations to extreme habitats due to its location in one of the world’s driest regions, comprised primarily of desert land cover (approximately 95%).9,10
The Makkah Region, located in the western part of Saudi Arabia, encompasses an area of approximately 153,128 km2 and exhibits a large degree of geographic and ecological variability.11 The region includes coastal plains bordering the Red Sea, the Hejaz Mountains, and many different sand and rock deserts. The geographic variability exhibited throughout the region creates a variety of soil environments with unique physicochemical properties, such as differences in soil moisture, pH, electrical conductivity, and mineral composition.12,13 It is expected that this environmental variability will create unique microbial communities that are adapted to their respective environments.14 However, despite the variability in soil environments within the Makkah Region, there has been a lack of comprehensive research examining the diversity of bacteria across these different soil environments.15,16
Molecular identification using the 16S ribosomal RNA (rRNA) gene has revolutionized the field of microbial ecology by enabling the identification and enumeration of microbial communities without requiring the prior cultivation of the organisms.17,18 The 16S rRNA gene contains highly conserved regions useful in designing universal primers and variable regions (primarily the V3-V4 regions), providing adequate phylogenetic resolution to differentiate between bacterial species.19 Sequencing technology, such as Illumina MiSeq, has greatly increased the number of sequences produced from environmental samples, thus allowing for complete characterization of both dominant and rare community members.20,21 Molecular identification coupled with the use of cultivation-based isolation techniques and biochemical characterization represents a comprehensive approach to investigating soil microbial communities. The combination of the discovery potential of molecular techniques with the functional information provided by culturable isolates represents a powerful method for studying soil microbial communities.22 The primary aims of this study were to isolate and characterize culturable bacterial strains from various soil types in the Makkah region; to identify isolated bacterial strains using morphological and molecular (16S rRNA gene sequence analysis); to measure the physicochemical properties of the soils; to analyze the relationships between soil properties and the structures of bacterial communities; and to contribute to our knowledge of microbial diversity in arid terrestrial ecosystems of the Arabian Peninsula.
Study areas
The soil-sampling initiative was conducted across all ten administrative provinces constituting the Makkah Region of the Kingdom of Saudi Arabia, situated in the western part of the country. The Makkah Region is an example of a coastal-inland macro ecoregion, where it extends from the shores of the Red Sea to the Hejaz escarpment and adjacent inland plateaus over an area of approximately 153,128 km2. The ten provinces selected for inclusion in this research were: Al-Qunfudhah, Mecca (the Fair Capital Governorate), Jamoum, Jeddah, Kamil, Khulais, Al-Laith, Rabigh, Taif, and Turbah. Each of these provinces was selected to represent a variety of geomorphic conditions; specifically, coastal, peri-coastal, and inland. Therefore, they can be used to illustrate the diversity in soil-forming processes found on land influenced by marine processes and on terrestrial processes. The spatial layout of this research effort has included marine-influenced plain areas of relatively low elevation, for example, Jeddah, Rabigh, and Al-Laith; Wadi/piedmont systems, for example, Khulais, Jamoum, and Kamil; and upland areas of the Hejaz Mountains or to the east of them, for example, Taif and Turbah. Collectively, these types of regions allow researchers to examine differences in climate (temperature) and hydrologic characteristics (intermittent precipitation and/or high salinity inputs due to both aerosols and soils), as well as variations in soil texture, which are common characteristics of arid environments found in western Arabia. A map illustrating the geographic location of sampling is presented in Figure 1. The centroid of the Mecca Region is located approximately at 21°30′ N Latitude and 41°00’E Longitude.
Figure 1. Map of Saudi Arabia showing the Makkah Region highlighted in red (Coordinates: 21°302 N, 41°002 E) (Source: mapsofIndia.com)
Sample collection and storage
A total of fifty soil samples, representative of a diverse range of edaphic conditions, were systematically collected from ten administrative provinces within the Makkah Region. The sampling methodology included three predominant soil texture classes: rocky (skeletal) soils, clay-dominated soils, and sandy soils, which are prevalent across both the western coastal areas and the inland regions of Saudi Arabia. Each soil sample was meticulously collected at a depth of 5-20 cm beneath the land surface using sterile instruments. This depth corresponds to the biologically active upper soil profile, recognized for its elevated microbial densities. Immediately upon collection, each sample was secured in a sterile plastic collection bag, accurately labeled with geographic coordinates and the date of collection, and subsequently transported on ice to the bacteriological laboratory within a 4 hrs window post-collection to prevent alterations in the microbial community composition. All samples underwent processing within 24 hrs of their arrival at the laboratory to ensure the integrity of the microbial analyses.
Physicochemical characterization of soil samples
Scanning Electron Microscopy (SEM) was used to characterize the soil samples based on the particle size distribution, morphology, surface texture, and elemental composition of individual particles. In order to conduct SEM analysis, the soil particles were mounted on aluminum stubs and then coated with a very thin layer of gold-palladium to increase the electrical conductivity and secondary electron emission of the sample. A JEOL 7500FA Scanning Electron Microscope (operational in Peabody, MA, USA) manufactured by JEOL LTD (Tokyo, Japan) was utilized to examine the samples, at an accelerating voltage of 10 kV, with a range of magnifications between 100× to 5000×, to visually record the particle shape, surface characteristics, and variations in color of the three major soil types. The pH of each soil type was determined utilizing a calibrated digital pH meter (model HI98107, Hanna Instruments, Woonsocket, RI, USA) and a 1:1 (w/v) ratio of soil to distilled water that had been allowed to equilibrate for thirty minutes at room temperature. The electrical conductivity (EC) of the soil types was also determined by measuring the EC of the same 1:1 (w/v) soil-water suspensions using a calibrated EC meter (Mettler Toledo-AG, Greifensee, Switzerland), and results are reported in mS/cm. The gravimetric measurement method was used to measure the soil moisture content (MC) of each soil type. Soil moisture content is defined as the weight of the soil before drying minus the weight of the dried soil, expressed as a percentage of the dry mass of the soil. This method is widely recognized as one of the most accurate methods available for determining soil moisture content. The three soil parameters examined were chosen because they have been previously identified as significant soil physical properties that influence both the composition and diversity of bacteria within arid soils.
Serial dilution and culture media
The processing of soil samples was conducted in alignment with established microbiological methodologies for the isolation of cultivable bacterial species. Specifically, each soil specimen (1 g) was suspended in 9 mL of sterile distilled water and subjected to vortex mixing to achieve a uniform initial suspension, corresponding to a 10–¹ dilution. Subsequent serial tenfold dilutions, ranging from 10-1 to 10-3, were conducted utilizing sterile distilled water as the diluent. From each dilution, aliquots of 1 mL were inoculated onto three distinct culture media designed to maximize bacterial diversity and facilitate the selective enrichment of specific microbial taxa: (1) nutrient agar, a general-purpose medium for cultivating heterotrophic bacteria; (2) mannitol salt agar, a selective medium aimed at isolating halotolerant and salt-tolerant bacteria, including certain Gram-positive cocci; and (3) MacConkey agar, which serves as both a selective and differential medium for Gram-negative, lactose-fermenting bacteria. Triplicate plates were prepared for each combination of soil sample and culture medium, followed by incubation under appropriate conditions.
Bacterial isolation techniques
A dual strategy was used to maximize the separation of viable bacteria from soil samples. First employed the soil dilution plate method was employed, which involved using the spread plate technique to place serial dilutions (10-1, 10-2, 10-3) of diluted soil samples on agar media, and placed the inoculated plates into an aerobic incubator for 24 hrs at 37 °C to allow the individual bacterial colonies to form and grow. The CFU/g (colony-forming unit/gram) of soil was determined for documentation. Secondly, we utilized the soil plate method to increase the recovery of various types of bacteria with different growth rates and spatial locations throughout the soil sample. This method involved placing 0.5 g of soil directly into a sterile petri dish containing 15 mL of sterile culture media that had been warmed to 45 °C to prevent premature gelification. The contents of the petri dish were gently mixed to distribute the soil evenly through the semi-solid agar matrix until it solidified. After solidification, the plates were incubated at 37 °C for 24 hours to allow the bacteria to grow. Following incubation, distinct colonies with varying morphologies, including color, size, transparency, pigmentation, and margin characteristics, were selected from each plate, and then individual bacterial isolates were streaked onto new nutrient agar plates to create pure cultures. Pure cultures were preserved in glycerol stocks at -20 °C for subsequent laboratory analysis.
Molecular identification via 16S rDNA sequencing
Genomic DNA was isolated from each pure bacterial isolate employing the InstaGene™ Matrix Genomic DNA Kit provided by Bio-Rad Laboratories (Hercules, CA, USA), following the protocol specified by the manufacturer. The isolated genomic DNA was utilized as the template for subsequent amplification of the 16S ribosomal DNA gene by PCR. The 16S rDNA region was subjected to amplification utilizing universal primer pairs designed to target conserved sequences that flank the hypervariable V4-V5 region. The primers employed were the forward primer 785-F (5′-GGATTAGATACCCTGGTA-3′) and the reverse primer 907-R (5′-CCGTCAATTCMTTTRAGTTT-3′). PCR reaction mixtures were prepared with a total volume of 20 µL, composed of final concentrations as follows: 1× Taq PCR buffer (10 mM Tris-HCl, 50 mM KCl, pH 8.3), 200 µM each of the deoxynucleotide triphosphates (dNTPs), 1.0 µM each of the forward and reverse primers (with a stock concentration of 10 pmol/µL), 0.5 U of KOMA Taq DNA polymerase (from a stock concentration of 2.5 U/µL), 20 ng of genomic DNA template, with HPLC-grade distilled water added to achieve the final reaction volume. The thermal cycling protocol implemented comprised the following parameters: an initial denaturation phase at 95 °C for 5 min; 30 amplification cycles incorporating denaturation at 95 °C for 30 sec, primer annealing at 55 °C for 2 min, and an extension phase at 68 °C for 1.5 min; concluded by a final extension step at 68 °C for 10 min. Verification of PCR amplicons, with an expected size of 1120 bp, was conducted via electrophoretic separation on 1% agarose gels stained with ethidium bromide. The bands were visualized using ultraviolet transillumination and compared against a 100-bp DNA ladder standard for size determination.
DNA sequencing and sequence analysis
Purified 16S rDNA PCR products underwent bidirectional sequencing employing the BigDye™ Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA) alongside the forward and reverse amplification primers. The cycle sequencing reactions were executed in an automated thermal cycler under the conditions delineated by the manufacturer. Following sequencing, extension products were subjected to ethanol precipitation to eliminate dye terminators, and subsequently analyzed using the ABI 3730xl DNA Analyzer (Applied Biosystems), an automated capillary sequencing system operated at Macrogen, Inc. (Seoul, South Korea). Forward and reverse sequences were processed and assembled into consensus sequences utilizing Geneious Prime software version 2020.1.2. Quality filtering was implemented to retain only bases with high-confidence (Phred quality score ≥Q20). The resultant consensus 16S rDNA sequences were compared against the comprehensive nucleotide sequence database hosted by GenBank using the Basic Local Alignment Search Tool (BLAST) with default parameters to ascertain the closest matching reference sequences and assign taxonomic classification. Bacterial isolates were identified to the genus level based on a sequence similarity of ≥97% to reference sequences catalogued in GenBank, with provisional species assignments determined in accordance with established taxonomic conventions pertinent to 16S rRNA based classification.
Secondary structure analysis of 16S rDNA sequences
The consensus 16S rDNA sequences underwent analysis to determine thermodynamically stable RNA secondary structures utilizing the RNAfold web server, hosted by the University of Vienna, Austria. This analysis employed the Turner thermodynamic model in conjunction with a dynamic programming algorithm. The minimum free energy (MFE) folding method was implemented to predict the secondary structure conformations with the lowest energetic cost for each sequence. The resulting predicted structures were represented in both dot-bracket notation and graphical diagrams, with particular emphasis placed on conserved structural domains, namely the 5′ central, 3′ major, and 3′ minor domains, as well as the hypervariable loop regions that are characteristic of bacterial 16S rRNA molecules. Gibbs free energy values (G, kcal/mol) were computed for each predicted structure as an indicator of thermodynamic stability. The secondary structures were then validated against reference models of 16S rRNA structures and compared across isolates derived from various soil types and phylogenetic lineages, aiming to discern correlations between structure and sequence.
Statistical analysis
Descriptive statistical analyses, including the computation of mean and standard deviation (SD), were conducted for all physicochemical parameters across soil samples categorized by province and soil type. Frequency distributions and enumeration of bacterial isolates were organized according to morphotype, preliminary identification category, and their definitive taxonomic classification based on 16S rDNA sequences. All statistical analyses and graphical representations of data were executed utilizing the Statistical Package for the Social Sciences (SPSS), version 23.0 (IBM Corp., Armonk, NY, USA). Descriptive statistical outcomes are expressed in terms of counts, percentages, and mean values accompanied by their standard deviations.
Soil physicochemical characteristics
The physicochemical characterization of soil samples obtained from the ten governorates within the Makkah Region demonstrated significant spatial variability in critical edaphic parameters, as depicted in Figure 2. The soil pH measurements indicated a range from 7.00 in the Kamil Governorate to 7.81 in the Khulais Governorate, consistent with the alkaline nature typical of soils found in arid regions of Saudi Arabia. The overall mean soil pH across all governorates was calculated to be 7.30 ± 0.29 (mean ± SD), where eight out of the ten governorates (Al-Qunfudhah, Fair Capital, Jamoum, Jeddah, Laith, Rabigh, Taif, and Torba) exhibited pH values within a narrow range of 7.10-7.40. In contrast, Kamil and Khulais Governorates showed significant deviations, with Khulais registering the highest pH value at 7.81, approximately 0.81 units above the regional mean. Soil moisture content (MC) displayed considerable spatial heterogeneity, spanning from a minimum of 0.50% in Fair Capital Governorate to a maximum of 12.0% in Jeddah Governorate. This represents a 24 fold variation, indicative of substantial differences in water retention capacity and local hydrological conditions. Coastal and peri-coastal governorates, such as Jeddah, Rabigh, and Al-Qunfudhah, exhibited elevated mean moisture content values of 9.23%, 4.50%, and 1.20%, respectively, in comparison to their inland and upland counterparts. Notably, Jeddah Governorate, situated directly along the Red Sea coast, significantly exceeded the regional mean moisture content of 4.52% ± 3.56%, likely due to the influence of maritime aerosol deposition and proximity to groundwater sources. Conversely, Fair Capital Governorate, encompassing the city center of Mecca at a high elevation within the interior Hejaz, recorded the lowest moisture content at 0.50%, reflecting its location in the region’s most extreme arid zone.
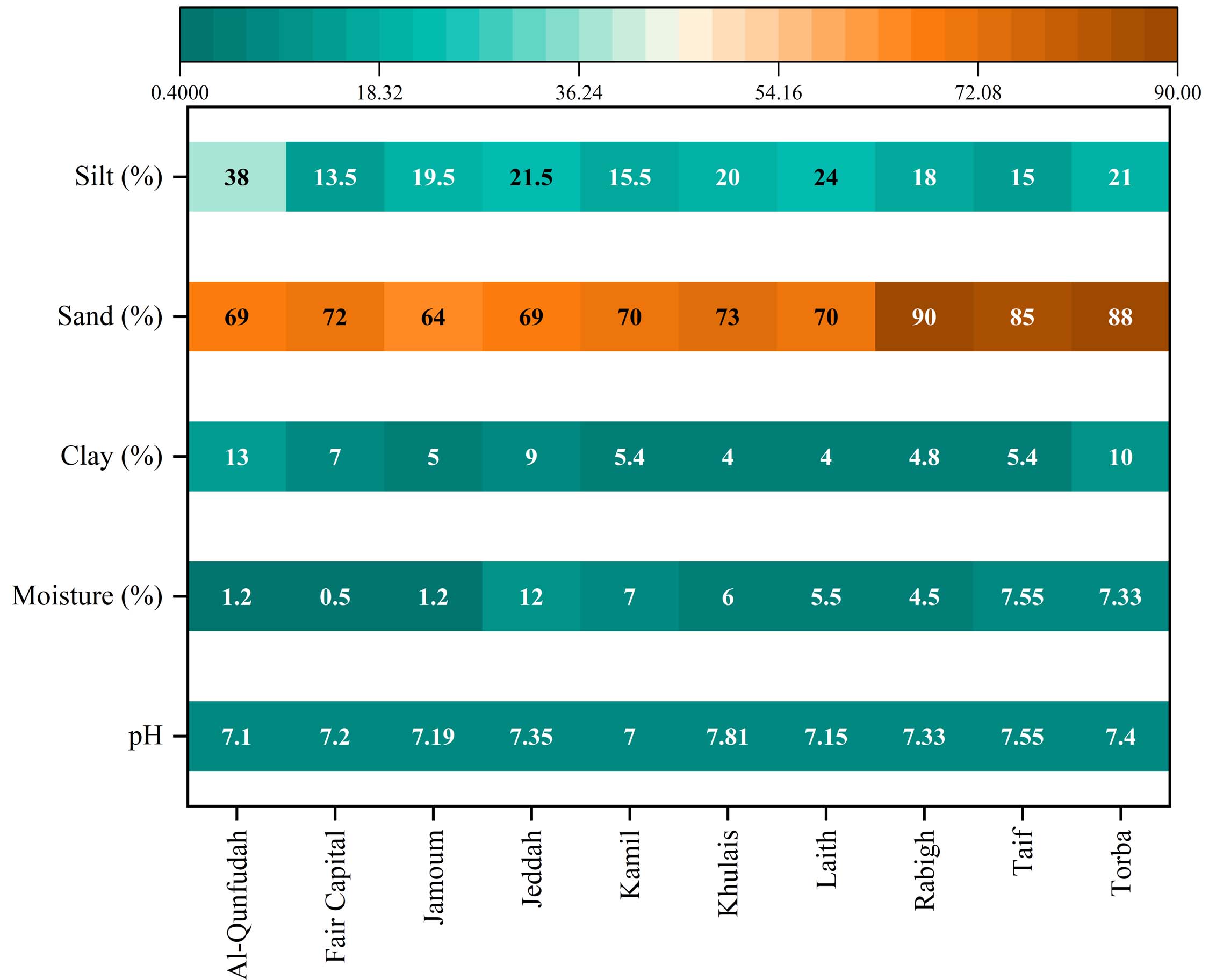
Figure 2. A comparative analysis of soil properties across various governorates within the Makkah region, which served as the sampling sites for this study
The analysis of soil particle size distribution across the ten governorates predominantly indicated sand-rich soil textures, which are typical of coastal and desert regions. The sand content varied from 64.0% in Jamoum Governorate to 90.0% in Rabigh Governorate, with an average of 74.8% ± 8.22% across all examined governorates. Except for Jeddah, all governorates exhibited sand percentages exceeding 69.0%, corroborating the prevalence of coarse-textured soils. Rabigh and Torba Governorates demonstrated the highest sand percentages (90.0% and 88.0%, respectively), indicative of extensive aeolian sand deposits and coastal dune systems in these littoral areas. The clay content was significantly lower, ranging from 4.0% in Khulais and Laith Governorates to 13.0% in Al-Qunfudhah Governorate, with an average of 6.94% ± 2.69%. Torba Governorate showed a relatively high clay content (10.0%) in comparison to other inland and southern governorates, likely due to wadi alluvial sedimentation. Silt content exhibited moderate variability, with values ranging from 13.5% in Fair Capital Governorate to 38.0% in Al-Qunfudhah Governorate, and an average of 20.5% ± 7.07%. Al-Qunfudhah Governorate, situated along the southwestern coastal plain, was distinguished by its high silt percentage (38.0%), suggesting significant contributions of fine-grained marine and deltaic sediments. The classification of soil texture using the USDA classification framework revealed that seven governorates, Fair Capital, Jamoum, Kamil, Khulais, Laith, Rabigh, and Taif, possess a loamy sand texture. Meanwhile, Al-Qunfudhah and Torba governorates are characterized by a sandy loam texture, attributed to their higher silt and clay content. In contrast, Jeddah Governorate exhibits a sandy clay loam texture, indicative of its relatively balanced ratios of sand, clay, and silt. These textural classifications illustrate a continuum from extremely coarse-textured aeolian and coastal sandy soils to finer-textured pedogenic soils, which are shaped by alluvial and marine depositional processes.
Bacterial abundance
The analysis of culturable bacterial isolates obtained from soil samples across ten governorates demonstrated significant spatial variability in bacterial abundance (Figure 3). The total number of bacterial isolates ranged from 6 in the Al-Qunfudhah Governorate to 20 in the Fair Capital Governorate, indicating a 3.33-fold variation in culturable bacterial density across different geographic regions. The cumulative count of bacterial isolates from all fifty soil samples (n = 50) across the ten governorates amounted to 100 isolates, resulting in an average abundance of 10.0 isolates per governorate. Notably, the Fair Capital Governorate, which includes the city of Mecca situated at a high elevation, exhibited the highest bacterial abundance with 20 isolates, constituting 20% of the total isolates. This represents the most extensive cultivable bacterial community identified from a single governorate. This elevated abundance is of particular interest considering Fair Capital’s notably low soil moisture content of 0.50%, suggesting that urban environmental factors and potential anthropogenic soil amendments, such as composted organic matter and wastewater irrigation, may augment cultivable bacterial populations despite arid conditions. In contrast, the Jeddah Governorate, positioned along the Red Sea coast, had the second-highest isolate count of 15 isolates, representing 15% of the total, which corresponds with its relatively higher soil moisture content of 12.0%, the highest among all governorates. Collectively, these two governorates, Fair Capital and Jeddah, accounted for 35% of the total bacterial isolates recovered. Moreover, the study documented intermediate levels of bacterial abundance, ranging from 8 to 9 isolates per governorate, constituting 8%-9% of the total isolates, in the governorates of Jamoum, Khulais, Laith, and Rabigh. In contrast, the governorates of Kamil, Torba, and Al-Qunfudhah reported relatively lower abundances, with 7, 7, and 6 isolates, respectively. Notably, Al-Qunfudhah Governorate exhibited the lowest bacterial abundance with 6 isolates, a finding that stands in stark contrast to its high silt content (38.0%) and moderate moisture level (1.20%). This discrepancy suggests that soil texture or other unassessed physicochemical parameters might be limiting the cultivable bacterial populations in this coastal southwestern region.
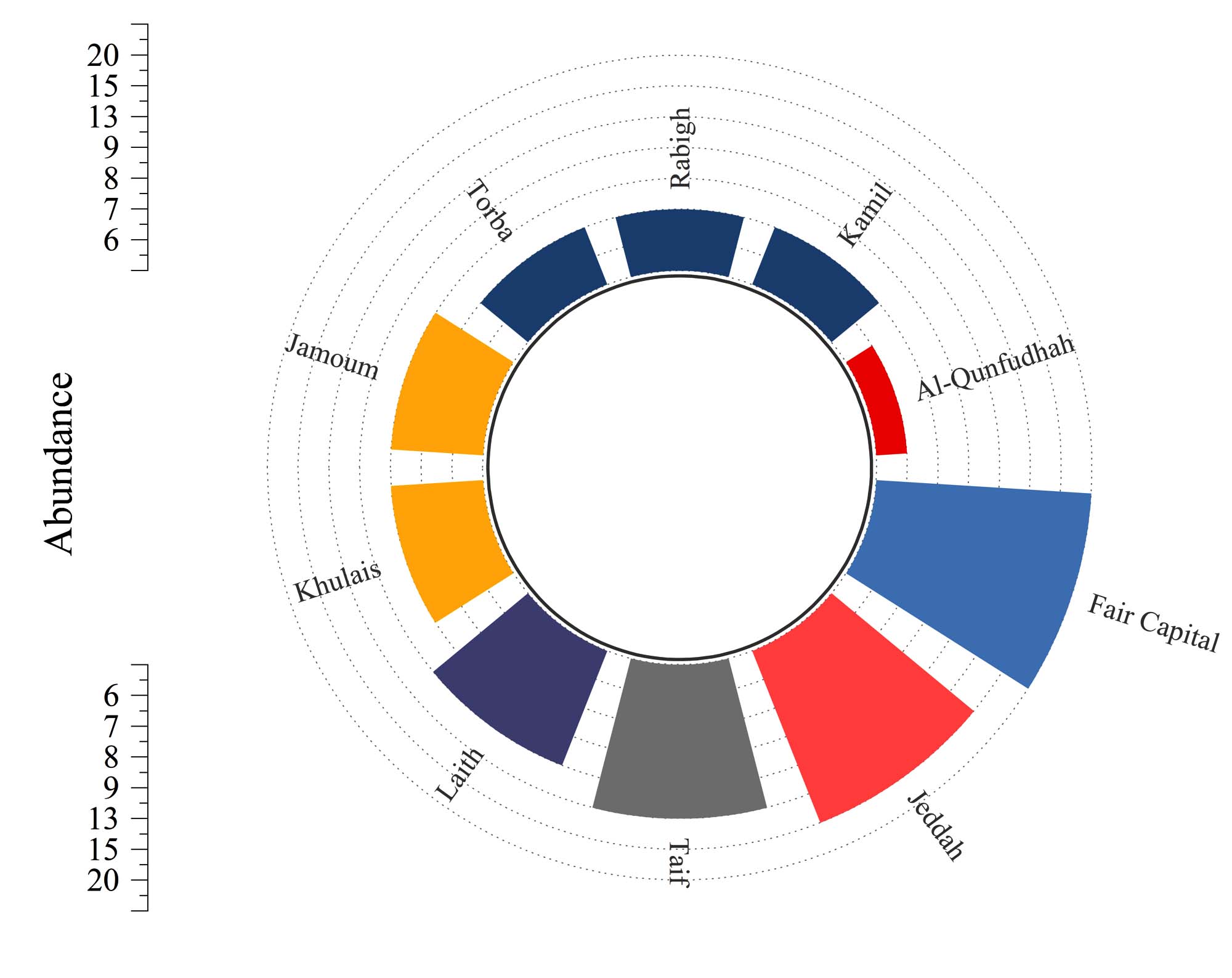
Figure 3. Distribution of bacterial populations among ten governorates in the Makkah region
In the PCA biplot (Figure 4), PC1 depicts a gradient ranging from clay- and silt-enriched soils at the negative end to sand-dominated soils at the positive end. Nonetheless, the link between soil texture and bacterial abundance is neither linear nor predictable based solely on texture. The strong positive loading of “Bacterial Abundance” along the positive PC1 axis indicates some degree of association between granulometric composition and microbial populations, yet this relationship shows marked variability among governorates. Sites with high PC1 scores host contrasting bacterial communities: Taif (85% sand) supports 13 bacterial isolates, whereas Rabigh (90% sand) and Torba (88% sand) each contain only 7, illustrating that a coarse texture by itself does not ensure favorable conditions for microbial colonization. Likewise, Fair Capital (72% sand) attains the highest bacterial abundance (20) despite exhibiting the lowest soil moisture (0.5%), underscoring that bacterial abundance cannot be attributed to soil texture or moisture in isolation. PC2 mainly contrasts fine-textured soils with bacterial abundance rather than reflecting soil texture alone, with clay and silt loading positively and bacterial abundance loading negatively. The close alignment of the “Silt” and “Clay” vectors signifies their joint contribution to soil fineness rather than an antagonistic relationship. Al-Qunfudah appears in the upper-left portion of the biplot, consistent with its high clay (13%) and silt (38%) contents and lowest bacterial abundance (6), suggesting that elevated proportions of fine particles may limit bacterial populations. This trend is not consistent across all sites: Jeddah has lower clay (9%) and comparable silt (21.5%) to Al-Qunfudah yet exhibits much higher bacterial abundance (15), indicating that clay and silt levels alone do not fully account for bacterial distribution. Fair Capital and Jeddah show the highest bacterial abundances (20 and 15, respectively), but their environmental profiles differ substantially. Jeddah is characterized by moderate sand (69%), relatively high moisture (12%), and moderate clay (9%), a balanced textural and moisture regime that appears conducive to robust microbial growth. In contrast, Fair Capital’s remarkably high bacterial abundance occurs despite extremely low moisture (0.5% versus 12% in Jeddah), implying that other unmeasured variables such as nutrient status, organic matter content, subsurface water retention, or specific microbial adaptations exert stronger control than texture or surface moisture alone. The extreme position of Fair Capital on the biplot therefore represents a key outlier that cannot be adequately explained within a conventional soil texture–moisture framework and merits additional investigation. Kamil, Laith, and Jamoum cluster near the center of the plot, reflecting intermediate textures (4%-5.4% clay, 15.5%-24% silt) and moderate bacterial abundances (7-9). These governorates appear to possess relatively balanced edaphic conditions that sustain baseline microbial populations without pronounced environmental extremes. Torba occupies a distinct location in the upper-right quadrant, with very high sand content (88%), elevated silt (21%), and moderate moisture (7.33%), yet only moderate bacterial abundance (7). Notably, Torba’s moisture level is similar to that of Taif (7.55%, 13 bacteria) and Kamil (7%, 7 bacteria), further indicating that moisture availability alone cannot explain differences in bacterial abundance. The pH vector shows weak loadings on both PC1 and PC2, and measured pH values fall within a narrow range (7.0-7.81; near-neutral to slightly alkaline) at all sites, suggesting that pH is not a primary factor structuring bacterial distribution in this dataset. Overall, contrasts among Rabigh and Torba (high sand, low bacterial abundance), Taif (high sand, moderate abundance), and Fair Capital (very high abundance at minimal moisture) indicate that spatial patterns of bacterial distribution are not chiefly driven by soil texture or moisture alone, but more likely reflect the influence of additional, unmeasured edaphic and biological determinants.
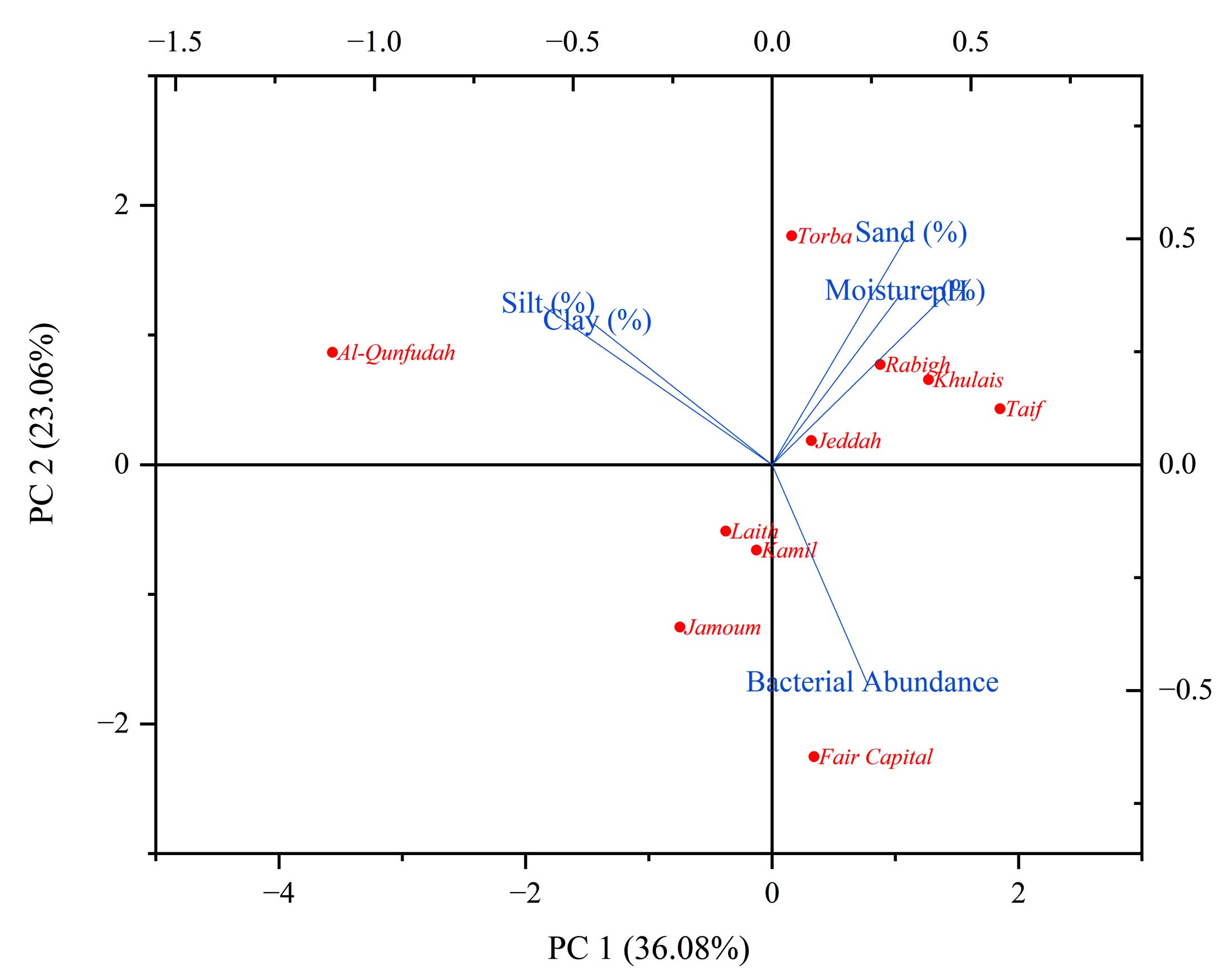
Figure 4. PCA biplot illustrating relationships between soil physicochemical properties and bacterial abundance across 10 Saudi Arabian governorates. Governorates are projected onto PC1 and PC2, explaining 59.14% of the total variance
SEM analysis of soil particle morphology and microbial communities
Scanning electron microscopic (SEM) examination at 100× magnification (scale bar = 500 µm) revealed substantial variation in soil particle size distribution, morphology, and surface colonization by microbial biofilms across the ten governorates of the Makkah Region (Figure 5). The SEM analysis provided direct visual documentation of the relationship between particle characteristics predicted by granulometric analysis and the actual morphological heterogeneity present in soil samples. Al-Qunfudhah Governorate (Panel A): SEM micrographs displayed a relatively heterogeneous particle size distribution dominated by medium to coarse sand grains (250-500 µm) intermixed with fine silt-clay aggregates. Particle surfaces exhibited moderate microbial colonization with visible biofilm formation, consistent with this governorate’s elevated silt content (38.0%) and intermediate bacterial isolate abundance (6 isolates). Particles displayed rounded to subrounded morphologies typical of marine-influenced coastal sediments subjected to wave action and abrasion. Fair Capital Governorate (Panel B): Micrographs displayed predominantly fine to medium sand grains (100-300 µm) with relatively sparse surface microbial colonization despite this governorate’s exceptionally high bacterial isolate abundance (20 isolates). Particle surfaces appeared relatively clean with minimal biofilm development, suggesting that culturable bacteria may occupy cryptic microsites (e.g., interstices between grains, subsurface pores) not visible in cross-sectional SEM views. This apparent discrepancy between high bacterial isolate recovery and limited visible biofilm suggests that the fair capital soil harbors predominantly planktonic or minimally biofilm-associated bacterial populations.

Figure 5. Presents SEM images illustrating the morphology of soil particles and the extent of microbial colonization on their surfaces, derived from ten governorates within the Makkah Region. The micrographs, captured at 100× magnification with a scale bar of 500 µm, depict the following locations: (A) Al-Qunfudhah, (B) Fair Capital, (C) Jamoum, (D) Jeddah, (E) Kamil, (F) Khulais, (G) Laith, (H) Rabigh, (I) Taif, and (K) Torba
Jamoum Governorate (Panel C): SEM micrographs revealed fine to medium sand particles (150-350 µm) with conspicuous spherical to subspherical microbial cells and biofilm aggregates distributed across particle surfaces and grain interstices. Extensive microbial colonization was visually apparent, with bacterial cells forming dense biofilm networks on multiple particle surfaces, consistent with this governorate’s moderate bacterial isolate count (8 isolates) and loamy sand soil texture. Jeddah Governorate (Panel D): Micrographs displayed medium sand grains (200-450 µm) with pronounced microbial biofilm formation, including visible clusters of cocci-like bacterial cells and filamentous structures characteristic of Actinobacteria or fungal hyphae. This extensive microbial colonization was consistent with Jeddah’s high soil moisture (12.0%), the highest among all governorates, and its substantial bacterial isolate abundance (15 isolates). The rich microbial biofilm networks observed in SEM images correlated with Jeddah’s coastal location and relative hydrologic favorability for microbial growth. Kamil Governorate (Panel E): SEM examination revealed predominantly medium sand particles (200-400 µm) with minimal biofilm colonization and relatively sparse visible microbial populations on particle surfaces. Particles appeared angular to subangular, reflecting limited weathering and sediment transport. This sparse visible biofilm correlated with Kamil’s moderate bacterial isolate count (7 isolates) and low soil moisture (7.00%). Khulais Governorate (Panel F): Micrographs displayed fine to medium sand particles (100-300 µm) with minimal visible microbial colonization on exterior particle surfaces. Particles exhibited well-rounded morphologies and relatively clean surfaces, despite this governorate’s moderate bacterial isolate abundance (8 isolates). Similar to Fair Capital, this observation suggests substantial populations of subsurface or interstitial bacteria not readily visible in cross-sectional SEM preparations. Laith Governorate (Panel G): SEM images revealed predominantly fine sand particles (100-250 µm) with sparse surface colonization by microbial biofilms. Particle surfaces appeared relatively weathered with etching patterns consistent with chemical dissolution and bacterial metabolic activity. Bacterial isolate count (9 isolates) and moisture content (5.50%) were moderate, suggesting environmental conditions were partially favorable for microbial growth.
Rabigh Governorate (Panel H): Micrographs displayed large coarse sand grains (300-600 µm) characteristic of this governorate’s elevated sand percentage (90.0%). Particle surfaces showed minimal visible biofilm formation and sparse microbial colonization, correlating with this governorate’s moderate bacterial isolate count (7 isolates) and intermediate moisture (4.50%). The prevalence of large sand grains and a limited fine-grained sediment matrix may have restricted microbial population densities. Taif Governorate (Panel I): SEM examination revealed medium to coarse sand particles (200-500 µm) with moderate visible biofilm colonization, including identifiable bacterial cell clusters and amorphous biofilm material. This moderate microbial colonization was consistent with Taif’s substantial bacterial isolate recovery (13 isolates) and elevated soil moisture (7.55%), representing upland environmental conditions influenced by the Hejaz escarpment. Torba Governorate (Panel K): Micrographs displayed heterogeneous particle sizes including coarse sand grains (300-600 µm) and identifiable clay-silt aggregates, consistent with this governorate’s elevated clay content (10.0%) and sandy loam texture. Pronounced biofilm development was visible, with extensive microbial colonization including bacterial cell clusters and filamentous structures. This rich microbial biofilm network correlated with Torba’s relatively high bacterial isolate abundance (13 isolates) and intermediate moisture (7.33%), suggesting that the presence of fine-grained sediments may enhance water retention and create favorable microsites for biofilm formation and microbial colonization.
Molecular identification and phylogenetic positioning
The multiple sequence alignment of partial 16S rRNA gene sequences (Figure 6) was conducted. The three Staphylococcus hominis isolates (PP758355.1, PP758356.1, PP758354.1) exhibited nearly identical 16S rRNA gene sequences across the aligned region, with sequence identity exceeding 99.5%. Detailed examination of the alignment identified only sporadic single-nucleotide polymorphisms (SNPs) at loci within hypervariable regions (e.g., positions 163, 383, 424), which is indicative of intraspecific strain-level variation. This notable sequence similarity corroborated that all three isolates belonged to the same bacterial species, S. hominis, and likely represented closely related strains originating from genetically similar source populations within the Makkah Region soil samples. The consensus sequence for S. hominis across these isolates exhibited 100% identity in conserved regions and >98% identity in variable regions when compared to the nearest GenBank reference sequences. The four Gram-negative bacterial isolates (Pseudomonas aeruginosa, Erwinia phyllosphaerae, Xanthomonas maliensis, and Dyella marensis), which represent distinct genera within the Proteobacteria phylum, showed substantial sequence divergence in hypervariable regions while maintaining high conservation in stem regions. Pairwise sequence identity calculations between isolate sequences and their closest GenBank matches ranged from 97.0%-99.8%, surpassing the conventional 97% threshold for species-level bacterial identification via 16S rRNA gene sequencing. The S. hominis isolates exhibited the highest sequence identity to reference strains (99.5%-99.8%), which is consistent with their classification as a well-characterized human-associated commensal species with extensive sequence representation in public databases. The environmental isolates (E. phyllosphaerae, X. maliensis, D. marensis) showed slightly lower but still robust sequence identities (97.5%-98.5%), reflecting their status as less extensively sequenced environmental taxa with greater intraspecific genetic diversity.

Figure 6. Multiple Sequence Alignment of 16S rRNA Gene Sequences from Seven Representative Bacterial Isolates and Closest GenBank Reference Sequences
A rooted phylogenetic tree derived through neighbor-joining (NJ) analysis of 16S rRNA gene sequences elucidated the evolutionary placement of the seven predominant bacterial isolates obtained from the soils of the Makkah Region, in relation to their closest validly published reference strains archived in the NCBI GenBank nucleotide database (Figure 7 and Table 1). The topology of the tree delineated two principal clades, which broadly correspond to the two primary bacterial phyla represented within the isolate collection: (1) a Firmicutes clade encompassing the three Staphylococcus hominis isolates (PP758355.1, PP758356.1, and PP758354.1), and (2) an expansive Proteobacteria clade comprising the four Gram-negative isolates (Pseudomonas aeruginosa NR_113169.1, Erwinia phyllosphaerae NR_041323.1, Xanthomonas maliensis NR_113599.1, and Dyella marensis NR_179668.1). This binary clade architecture accurately mirrored the fundamental phylogenetic bifurcation of culturable bacteria, thereby affirming the reliability of the sequence-based phylogenetic inference. The three isolates of Staphylococcus hominis exhibited a high degree of genetic similarity, as evidenced by their clustering with exceedingly short internodal branch lengths, with an evolutionary distance of less than 0.005 substitutions per site. This observation suggests near-identical sequences of the 16S rRNA gene, indicative of recent common ancestry or clonal proliferation originating from a single progenitor population. The close clustering of these isolates within a monophyletic clade, distinct from other Staphylococcus reference strains, corroborated their species-level identification. This finding implies that these isolates are likely independent derivations of the identical bacterial species, potentially sourced from the same or proximate soil sample locations. The bootstrap support values at the S. hominis clade node were exceedingly high, exceeding 99%, though not explicitly labeled in the figure, which is standard for such closely related groupings. This provides compelling statistical evidence for the monophyletic association of these three isolates. Within the expansive Proteobacteria clade, distinct generic subdivisions were elucidated with sufficient phylogenetic resolution. The Pseudomonas aeruginosa isolate (NR_113169.1) exhibited a separate branching pattern with moderate evolutionary distance from plant-associated Proteobacteria, such as the Erwiniaceae and related taxa, aligning with the deeper phylogenetic divergence observed between Pseudomonadales and Enterobacteriales. The Erwinia phyllosphaerae isolate (NR_041323.1) was grouped with other gamma-proteobacterial enterobacterial relatives, whereas Xanthomonas maliensis (NR_113599.1) demonstrated a branching characteristic that is consistent with its classification within the Xanthomonadaceae family. The Dyella marensis isolate (NR_179668.1) was identified as a distinct lineage within beta-proteobacteria sensu lato, exhibiting significant evolutionary distance from all other isolates, thus representing the most phylogenetically divergent taxon within the examined collection.
Table (1):
NCBI nucleotide BLAST identification results for all analyzed samples
Sample |
Blast result |
Cover % |
Ident. % |
E-value |
Acc. No. |
|---|---|---|---|---|---|
A3 |
Staphylococcus hominis |
100 |
99.81 |
0.00 |
PP758354.1 |
E2 |
Staphylococcus hominis |
100 |
99.37 |
0.00 |
PP758355.1 |
E3 |
Staphylococcus hominis |
100 |
99.36 |
0.00 |
PP758356.1 |
F5 |
Dyella marensis |
99 |
99.00 |
0.00 |
PP758357.1 |
W3 |
Pseudomonas aeruginosa |
100 |
90.22 |
0.00 |
PP758392.1 |
F6 |
Xanthomonas maliensis |
100 |
99.55 |
0.00 |
PP758377.1 |
S13 |
Erwinia phyllosphaerae |
100 |
98.39 |
0.00 |
PP758378.1 |
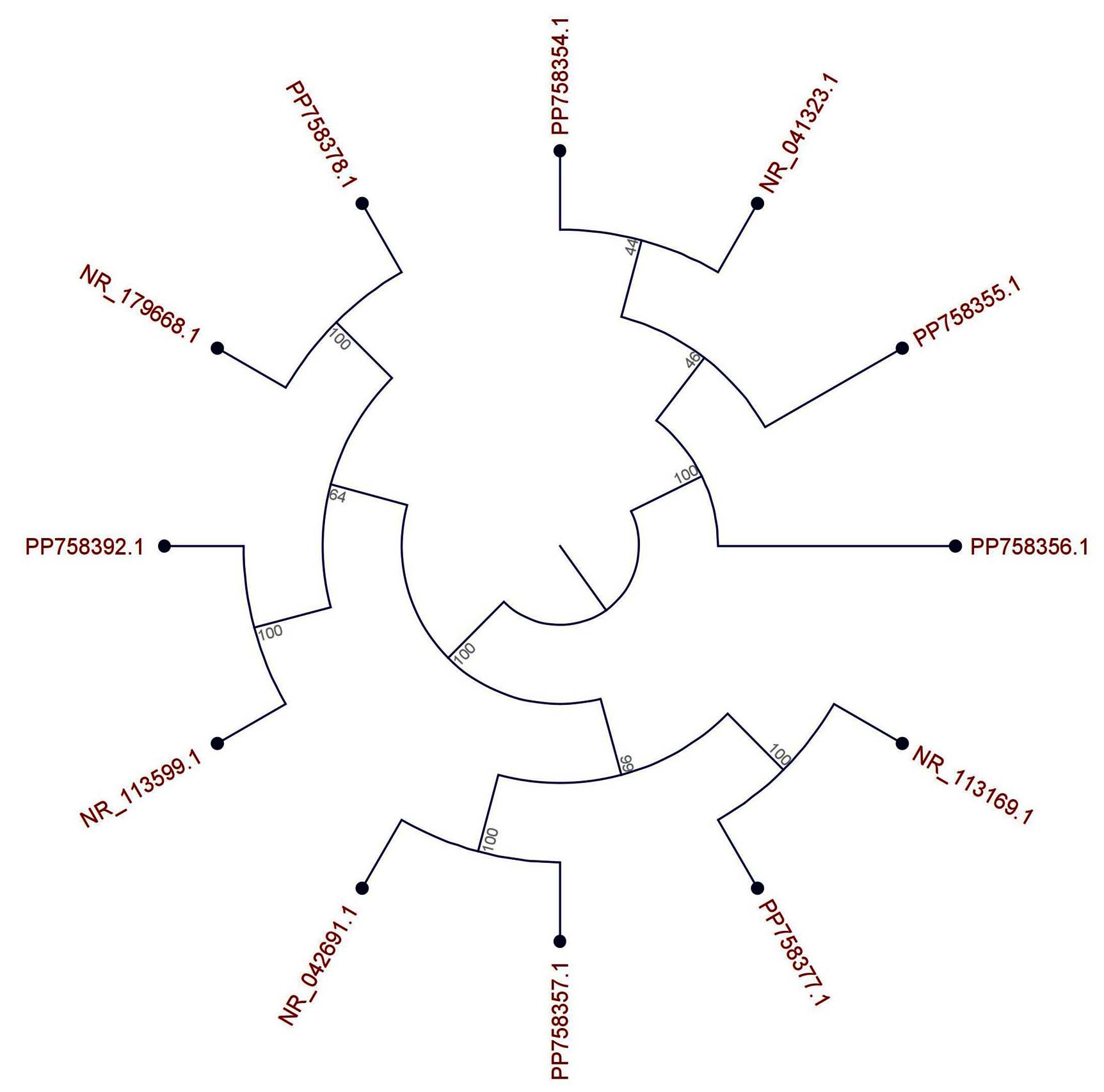
Figure 7. Neighbor-Joining Phylogenetic Tree of 16S rRNA Gene Sequences from Seven Predominant Bacterial Isolates and Reference Strains
Secondary structure fold analysis
The analysis of predicted RNA secondary structures for bacterial isolates and their corresponding reference sequences revealed significant inter-species structural divergence, with concurrent intraspecific structural conservation (Figure 8). This indicates that the architecture of the 16S rRNA secondary structure both reflects and reinforces phylogenetic relationships as determined by primary sequence analysis. A comparative investigation of the reference and isolate 16S rRNA secondary structures uncovered distinct variations in multi-branch loop (MBL) topology and stem organization. In the pair NR_179668 and PP758378, the reference structure was characterized by a large, open, and symmetrical central multi-branch loop connecting four major stems, whereas the isolate exhibited a highly constrained and elongated architecture dominated by a single long stem. This suggests a transition from a symmetrical to a constrained/asymmetrical MBL topology. The comparison between NR_042691 and PP758357 showed that while the reference structure possessed a large, open central MBL, the isolate’s MBL was smaller and more compact, with helices converging more tightly, indicating reduced accessibility at the central junction. For the NR_113599 and PP758392 pair, the reference structure contained a distinctive, large, and complex terminal multi-branch loop at the 3′ end, which was absent in the isolate, where the 3′ region folded into a simpler hairpin capped by a short stem. Conversely, the NR_113169 and PP758377 pair demonstrated near-perfect structural conservation, with both maintaining a large fan-like MBL connecting five major stems, showing no detectable variation in motif organization. Lastly, comparisons involving NR_041323, PP758354, PP758355, and PP758356 revealed more pronounced architectural divergence. The reference NR_041323 displayed an open and unconstrained central MBL, whereas PP758354 showed a tightly packed MBL incorporating a potential pseudoknot-like interaction near the base of the structure. The PP758355 isolate exhibited a highly linear conformation dominated by a single, continuous stem with minimal branching, contrasting with PP758356, which presented a more globular architecture with a distinct MBL connecting multiple stems. Collectively, these observations underscore the variability in MBL morphologies across isolates, ranging from open and symmetrical to constrained and stem-dominant conformations, with pseudoknot-like interactions emerging as a distinguishing feature in select isolates.

Figure 8. Comparative Secondary Structure Fold Analysis of 16S rRNA
The examination of the ensemble dynamics depicted by mountain and positional entropy plots (Figure 9). Mountain plots, integrating MFE, partition function (PF), and centroid profiles, revealed that sequences PP758378, NR_113599, NR_113169, and PP758377 exhibited highly aligned structural peaks (positions 500-1000), indicating stable ensembles with MFE conformations representative of the dominant structural state. Conversely, PP758392 and PP758354 displayed significant divergence between MFE and PF/centroid profiles, especially in the 3′ region (positions 1000-1500), suggesting that their MFE folds correspond to low-probability conformations within structurally diverse ensembles. Positional entropy analyses further demonstrated low entropy (<1.0 bit) in core stem regions, aligning with high structural stability, and elevated entropy (up to 3.0 bits) in terminal and loop regions, reflecting increased flexibility. The pronounced entropy spikes in the 3′ regions of PP758392 and PP758354 correspond to structural hotspots exhibiting conformational switching, suggesting potential roles in dynamic regulation and functional adaptation.
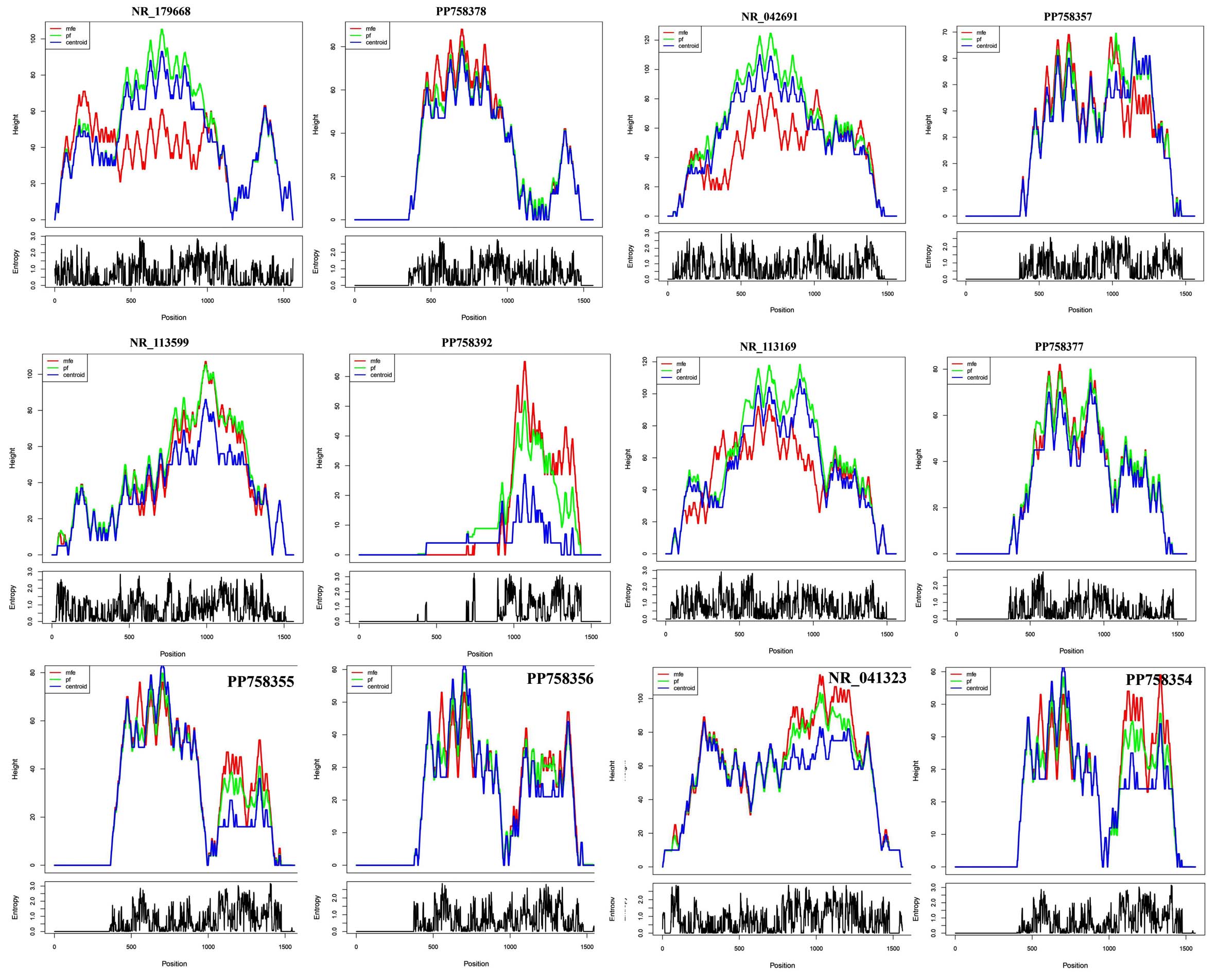
Figure 9. Analysis of RNA Structural Ensemble and Positional Entropy. The figure displays mountain plots (top) and positional entropy plots (bottom) for the RNA sequences. The mountain plots compare the height (structural depth) of the Minimum Free Energy (MFE, red), Partition Function (PF, green), and Centroid (blue) structures. The entropy plots show the positional entropy (in bits) for each nucleotide, indicating structural uncertainty
Sequences polymorphism
The investigation of single-nucleotide polymorphisms (SNPs), as illustrated in Table 2, elucidates disparities in evolutionary divergence from reference sequences, genomic mutation rates, and polymorphism frequencies at the population level. Although all three Staphylococcus hominis isolates exhibited more than 99.5% similarity in their 16S rRNA sequences, they demonstrated significant variability in SNP burden. S. hominis A3 (PP758354) was characterized by the lowest SNP count, with three substitutions occurring at nucleotide positions 12, 49, and 1042. Specifically, positions 12 and 49 revealed Γ→T and AΓ→T transversions, respectively, whereas position 1042 manifested an A→G transition. This SNP configuration yielded a nucleotide divergence rate of 0.29%, indicative of near-perfect sequence conservation and minimal intraspecific variation. Conversely, S. hominis E2 (PP758355) harbored four SNPs concentrated within the initial 60 nucleotides (positions 14, 39, 53, and 57), comprising three transversions (A→T, C→G, and G→T) and a single transition (G→A). The predominance of transversions (75%) is notable, as it inverts the typical prokaryotic pattern where transitions generally outnumber transversions by a ratio of approximately 2:1, implicating potential selective pressure or an atypical mutational bias in this isolate. Furthermore, S. hominis E3 (PP758356) exhibited the highest SNP count among the isolates, with seven substitutions (positions 24, 38, 476, 867, 972, 1068, 1086, and 1087), consisting of four transversions (C→A, G→T, T→G, A→C) and three transitions (T→C, A→G, A→G). The balanced 4:3 ratio (57% transversions) more closely aligns with general prokaryotic mutation paradigms. The extensive positional distribution of SNPs (24-1087) implies dispersed rather than clustered mutational events, and the nucleotide divergence rate of 0.70% suggests greater evolutionary distance from the reference sequence in comparison to E2 (0.40%) or A3 (0.29%). Meanwhile, Dyella marensis F5 (PP758357) demonstrated the highest SNP count overall, with eleven substitutions ranging from positions 8 to 1090. The SNPs included six transitions (A→G at 289, 682, 954, 1068; T→C at 644, 971; C→T at 1090) and five transversions (A→T at 8, 1067; A→C at 23; T→G at 1069), yielding a transition/transversion ratio of 1.2:1 (55% transitions), which is consistent with prokaryotic mutational trends. The extensive positional distribution and a nucleotide divergence rate of 0.99% imply the presence of geographically widespread polymorphisms and a potential ongoing genomic diversification or admixture among Dyella lineages. Similarly, Xanthomonas maliensis F6 (PP758377) demonstrated the presence of five SNPs at positions 50, 52, 58, 96, and 97. These SNPs consisted of three transitions (A→G at position 52; C→T at 96; C→G at 97) and two transversions (A→C at 50; T→G at 58). Notably, positions 96 and 97 are contiguous, forming a localized mutational cluster indicative of a potential hotspot or a short-region divergence. The resulting nucleotide divergence rate of 0.50% was intermediate among non-Staphylococcus isolates. Erwinia phyllosphaerae S13 (PP758378) exhibited the lowest SNP count among non-Staphylococcus isolates, with only two transversions (A→C at position 51 and A→G at position 114). Although this corresponded to a divergence rate of 0.80%, the limited sequence coverage (~250 nucleotides) likely underestimates the overall variation, yet it still reflects a close phylogenetic relatedness to the reference Erwinia sequence. Lastly, Pseudomonas aeruginosa W3 (PP758392) displayed the highest absolute SNP count, with fourteen substitutions predominantly distributed within the first 160 nucleotides (positions 25-153). These consisted of five transitions (C→T at 25 and 77; A→G at 34; G→C at 114) and nine transversions (T→A at 35; C→G at 60 and 139; G→C at 65; A→T at 82 and 126; T→G at 88; A→C at 102; G→T at 153), yielding a transversion-skewed ratio of 1.8:1 (64% transversions). The clustering of SNPs within this early sequence region proposes a mutational hotspot or a locus under diversifying selection.
Table (2):
SNPs identified in the seven bacterial isolates alongside their respective genomic positions, in comparison to homologous sequences obtained from the database
| Query Organisms | Reference | Position | SNPs | Type | ||
|---|---|---|---|---|---|---|
| Staphylococcus hominis A3 (PP758354) | Staphylococcus hominis (NR041323) | 12 | G | → | T | Transversion |
| 49 | A | → | T | Transversion | ||
| 1042 | A | → | G | Transition | ||
| S. hominis E2 (PP758355) | 14 | A | → | T | Transversion | |
| 39 | C | → | G | Transversion | ||
| 53 | G | → | T | Transversion | ||
| 57 | G | → | A | Transition | ||
| S. hominis E3 (PP758356) | 24 | C | → | A | Transversion | |
| 38 | G | → | T | Transversion | ||
| 476 | T | → | C | Transition | ||
| 867 | G | → | T | Transversion | ||
| 972 | T | → | G | Transversion | ||
| 1068 | A | → | G | Transition | ||
| 1086 | A | → | C | Transversion | ||
| 1087 | T | → | A | Transversion | ||
| Dyella marensis F5 (PP758357) | Dyella marensis (NR042691) | 8 | A | → | T | Transversion |
| 23 | A | → | C | Transversion | ||
| 289 | A | → | G | Transition | ||
| 644 | T | → | C | Transition | ||
| 682 | A | → | G | Transition | ||
| 954 | A | → | G | Transition | ||
| 971 | T | → | C | Transition | ||
| 1067 | A | → | T | Transversion | ||
| 1068 | G | → | A | Transition | ||
| 1069 | T | → | G | Transversion | ||
| 1090 | C | → | T | Transition | ||
| Xanthomonas maliensis F6 (PP758377) | Xanthomonas maliensis (NR113169) | 50 | A | → | C | Transversion |
| 52 | A | → | G | Transition | ||
| 58 | T | → | G | Transversion | ||
| 96 | C | → | T | Transition | ||
| 97 | C | → | G | Transversion | ||
| Erwinia phyllosphaerae S13 (PP758378) | Erwinia phyllosphaerae (NR179668) | 51 | A | → | C | Transversion |
| 114 | A | → | G | Transition | ||
| Pseudomonas aeruginosa W3 (PP758392) | Pseudomonas aeruginosa (NR113599) | 25 | C | → | T | Transition |
| 34 | A | → | G | Transition | ||
| 35 | T | → | A | Transversion | ||
| 60 | C | → | G | Transversion | ||
| 65 | G | → | C | Transversion | ||
| 77 | C | → | T | Transition | ||
| 82 | A | → | T | Transversion | ||
| 88 | T | → | G | Transversion | ||
| 102 | A | → | C | Transversion | ||
| 114 | G | → | C | Transversion | ||
| 126 | A | → | T | Transversion | ||
| 129 | C | → | G | Transversion | ||
| 139 | C | → | G | Transversion | ||
| 153 | G | → | T | Transversion | ||
Additionally, the identified insertions and deletions (InDels) provide compelling evidence of sequence assembly dynamics, structural rearrangements impacting the organization and secondary structure topology of the 16S rRNA gene (Table 3). In Staphylococcus hominis A3 (PP758354), a solitary InDel detected a thymine deletion (T-) at position 1061, situated within the terminal 3′ region corresponding to the V6 loop domain. This event signifies the loss of one nucleotide relative to the reference sequence and may result in local loop shortening or altered secondary structure topology. The minimal InDel burden, in conjunction with the modest SNP count (3 SNPs), indicates a relatively stable genomic architecture with minimal structural rearrangement. In contrast, S. hominis E2 (PP758355) demonstrated three total InDels, comprising two adjacent guanine insertions (G+ at positions 1083 and 1095) and two deletions (G- at position 26 and T- at position 1106). The insertions, concentrated within a 12-nucleotide span at the 3′ terminus, likely indicate a localized insertion or tandem duplication event extending the V6 loop by two nucleotides. Meanwhile, the spatially separated deletions suggest multiple independent mutational occurrences rather than a single concerted rearrangement. Furthermore, S. hominis E3 (PP758356) exhibited only one InDel, a guanine deletion (G-) at position 11 near the 5′ terminus, potentially representing either a sequencing artifact or authentic trimming of the ribosomal RNA. Despite possessing the highest SNP count among the Staphylococcus isolates (8 SNPs), this strain’s minimal InDel occurrence suggests that base substitutions rather than insertions or deletions dominate its sequence variation pattern. Similarly, Dyella marensis F5 (PP758357) contained two InDels, both insertions (C+ and G+) occurring at adjacent positions (1070 and 1078) within the 3′ V6 loop region. These events collectively extended the terminal loop by two nucleotides relative to the reference sequence. When coupled with its high SNP burden (11 SNPs, 0.99% divergence), these clustered insertions suggest marked sequence divergence from the reference Dyella strain, potentially indicative of a genetically distinct subspecies or population variant. Xanthomonas maliensis F6 (PP758377) exhibited only a single thymine deletion (T-) at position 12 in the 5′ region, indicating limited structural variation. Combined with its moderate SNP load (5 SNPs), this finding supports a relatively conserved genomic organization in this isolate. While the bacterial strain Erwinia phyllosphaerae S13 (PP758378) exhibited two insertion-deletion events (InDels): a loss of guanine (G-) at nucleotide position 14 and an addition of thymine (T+) at position 1101. Notably, the deletion at the 5′-terminus replicates a pattern previously identified in both Xanthomonas F6 and Staphylococcus hominis E3, hence suggesting the presence of genuine biological variation at the sequence extremities, or a persistent bias in 5′ sequencing coverage. The insertion event of thymine near the 3′-terminus further supports the hypothesis of independent mutational occurrences at both ends of the sequence. In contrast, the strain Pseudomonas aeruginosa W3 (PP758392) demonstrated the highest burden of InDel variations, comprising seven events in total: six insertions (G+, A+, T+, A+, T+, and T+ at positions 20, 27, 36, 37, 50, and 155, respectively) and a single deletion (T- at position 148). Of particular interest is that five out of these seven events (constituting 71%) were localized within a compact 30-nucleotide segment (positions 20-50), which includes two consecutive single-nucleotide insertions (T+ and A+ at positions 36 and 37), suggesting a localized mutational hotspot or a structurally unstable region.
Table (3):
InDels were detected in the seven bacterial isolates, and their positions were compared to similar ones retrieved from the database
| Query Organisms | Reference | Position | REF (hit) | ALT (query) | Type |
|---|---|---|---|---|---|
| Staphylococcus hominis A3 (PP758354) | Staphylococcus hominis (NR041323) | 1061 | T | – | Deletion |
| S. hominis E2 (PP758355) | 26 | G | – | Deletion | |
| 1083 | – | G | Insertion | ||
| 1095 | – | G | Insertion | ||
| 1106 | T | – | Deletion | ||
| S. hominis E3 (PP758356) | 11 | G | – | Deletion | |
| Dyella marensis F5 (PP758357) | Dyella marensis (NR042691) | 1070 | – | C | Insertion |
| 1078 | – | G | Insertion | ||
| Xanthomonas maliensis F6 (PP758377) | Xanthomonas maliensis (NR113169) | 12 | T | – | Deletion |
| Erwinia phyllosphaerae S13 (PP758378) | Erwinia phyllosphaerae (NR179668) | 14 | G | – | Deletion |
| 1101 | – | T | Insertion | ||
| Pseudomonas aeruginosa W3 (PP758392) | Pseudomonas aeruginosa (NR113599) | 20 | – | G | Insertion |
| 27 | – | A | Insertion | ||
| 36 | – | T | Insertion | ||
| 37 | – | A | Insertion | ||
| 50 | – | T | Insertion | ||
| 148 | T | – | Deletion | ||
| 155 | – | T | Insertion |
Soil physicochemical characterization and bacterial abundance
The physicochemical analysis of soil from the ten governorates of the Makkah Region exhibited notable spatial variability in critical edaphic parameters. The alkaline pH values observed (7.00-7.81) and the significant variation in moisture content (0.50%-12.0%) underscore the transitional gradient between coastal and inland areas, as well as the pronounced aridity of the region. These pH values align with the established characteristics of arid soils in Saudi Arabia, where calcium carbonate accumulation and limited acid-producing biological activity contribute to prevailing alkalinity.23,24 The pronounced moisture content gradient (0.50%-12.0%, with Jeddah displaying 24-fold higher moisture than Fair Capital) provides critical insight into the environmental heterogeneity selecting for distinct bacterial populations. This moisture heterogeneity aligns with reported patterns in arid soil microbiomes, where soil relative humidity represents one of the strongest predictors of bacterial diversity and composition.25,26 This observation supports the established principle that water availability, mediated through soil moisture content, represents a fundamental constraint on bacterial population sizes in arid environments.27 Notably, Fair Capital’s exceptionally high bacterial isolate count (20 isolates, 20% of total) despite minimal moisture (0.50%) suggests that anthropogenic modification (proximity to Mecca city, potential organic matter amendments, wastewater irrigation) may partially decouple the expected moisture diversity relationship in urban soil environments.28
The classification of soil texture indicated a predominance of sand-rich soils, with an average composition of 74.8%, and comparatively minor contributions from clay (6.94%) and silt (20.5%). Comparative analyses from global studies on arid soils reveal that bacterial diversity exhibits an inverse relationship with sand content.5 These correlations highlight the physical mechanisms wherein coarse sand particles afford limited water retention and reduced surface area for microbial colonization and nutrient retention. In contrast, finer-grained sediments enhance aqueous microhabitat stability and nutrient availability by increasing cation exchange capacity.29 This finding suggests that in the context of the Makkah Region’s extreme aridity and limited variation in texture (with sand content consistently over 60%), the influence of texture on moisture and nutrient availability may be partially mitigated by other environmental factors, particularly the absolute availability of moisture and the impacts of urban anthropogenic activities.30
The observed spatial heterogeneity, characterized by a 3.33-fold variation between the maximum values recorded in Fair Capital (20 isolates) and the minimum in Al-Qunfudhah (6 isolates), illustrates a geographic patterning that aligns with established determinants of microbial heterogeneity in arid soils.31 These determinants include proximity to aquatic sources, such as the Red Sea coast at Jeddah, urban anthropogenic enrichment, exemplified by Mecca city in the Fair Capital, and soil-forming processes, particularly the alluvial wadi sediments found in central governorates. The observed recovery of bacterial isolates from coastal and peri-coastal regions (with a combined mean of 9.3 isolates in Jeddah, Rabigh, and Al-Qunfudhah) compared to the interior highlands (Taif and Fair Capital with a mean of 10.5 isolates) presents a pattern found in other desert coastal ecosystems. In such systems, maritime aerosol deposition enhances the availability of salts, moisture, and nutrients compared to inland arid regions.32,33 Notably, the unexpectedly high bacterial abundance documented in Fair Capital despite its location in an extremely arid microenvironment indicates substantial modification of the aridity-diversity relationship by urban-anthropogenic influences. Urban soil environments are often subject to nutrient enrichment through waste streams, anthropogenic organic amendments (such as compost and food waste), and wastewater irrigation, which may mitigate moisture-related limitations on bacterial abundance.34 This phenomenon is corroborated by previous comparative studies, which have documented that anthropogenic soil alterations and urban management practices can significantly enhance cultivable bacterial populations, even in otherwise inhospitable environments.35
Molecular identification and phylogenetic insights
The analysis of the 16S rRNA gene sequencing elucidated a bacterial isolate collection that is predominantly comprised of seven species, specifically: three strains of Staphylococcus hominis, one Pseudomonas aeruginosa, one Erwinia phyllosphaerae, one Xanthomonas maliensis, and one Dyella marensis. This assemblage reflects the typical phylogenetic architecture of arid soil microbiomes, which are globally characterized by the dominance of Actinobacteria, Proteobacteria, and Firmicutes.5,36 The isolation of Pseudomonas aeruginosa is consistent with established reports of this widespread Gamma-proteobacterium in various soil ecosystems, including arid and desert terrains, where it is capable of enduring desiccation through mechanisms such as biofilm formation and pigment synthesis.37 The presence of plant-associated Enterobacteriales, specifically Erwinia phyllosphaerae and Xanthomonas maliensis, likely indicates the ecological influence of residual vegetation or plant detritus in the sampled substrates or may represent the oligotrophic persistence of plant pathogenic lineages within soil reservoirs.38 Furthermore, the detection of Dyella marensis, a gamma-proteobacterium (Family: Rhodobacteraceae), constitutes a noteworthy discovery, as it has been rarely reported and appears limited to specific soil environments.39
Whereas Staphylococcus hominis is a coagulase-negative staphylococcal species (CoNS) that is primarily associated with the human skin microbiome, where it typically occurs as a commensal organism with opportunistic pathogenic potential.40,41 Its recovery from soil thus warrants interpretation within an anthropogenic framework, as its pronounced host specificity renders direct environmental introduction from humans or secondary contamination highly plausible. Within clinical microbiology, CoNS, including S. hominis, are frequently regarded as contaminants in the absence of compelling etiological evidence, highlighting the necessity for cautious evaluation when these taxa are identified in extra-host or environmental matrices.42 Studies of the built environment demonstrate that S. hominis is capable of persisting outside the human host in human-occupied settings. For instance, S. hominis has been reported as a dominant aerobic taxon in the air microbiome of dental clinics, comprising approximately 22%-24% of recovered bacterial sequences.43 These observations indicate that skin-associated bacteria can become aerosolized, settle onto surfaces and particulate matter such as dust, and subsequently be transported to outdoor environments, where they may enter urban soils through atmospheric deposition and resuspension processes. Investigations of hospital wards further document environmental surface contamination by S. hominis, reinforcing its capacity to persist in anthropogenically modified habitats.44 Beyond human-derived sources, S. hominis has also been isolated from livestock and arthropod hosts, including bovine body sites and the microbiome of the malaria vector Anopheles stephensi.45 These findings broaden the range of potential inputs into soil, particularly in peri-urban or mixed-use landscapes characterized by the intersection of human activity, livestock production, and insect vector populations. Consequently, the detection of S. hominis in soil is more plausibly interpreted as reflecting a composite signal of human and non-human inputs rather than stable, autochthonous soil residency. From an urban ecology standpoint, the occurrence of S. hominis in soil is most parsimoniously explained as an indicator of anthropogenic influence rather than as evidence that this species constitutes a native soil taxon. Its frequent association with healthcare environments, documented resistance phenotypes, and demonstrated ability to persist on abiotic surfaces collectively suggest introduction via human activities, including waste streams, airborne dust, or surface runoff.
The hierarchical organization of divergence in 16S rRNA secondary structure among the seven bacterial isolates demonstrated complete congruence with phylogenetic relationships determined through primary sequence analysis, thereby providing independent validation for both phylogenetic and structural inferences. The Staphylococcus hominis isolates exhibited remarkable conservation in secondary structure. This exceptional structural homogeneity has significant implications for validating species-level classification among these isolates, as such perfect intraspecific structural concordance is anticipated only under conditions of minimal evolutionary divergence. This pattern aligns with established principles of RNA structural evolution, whereby conserved functional domains (stems) undergo gradual lengthening that reflects the accumulation of sequence divergence, while the overall topology remains constrained by ribosomal function.46 Previous studies on 16S rRNA secondary structure variation in cyanobacteria similarly reported that secondary structure variation provides a phylogenetic signal that complements primary sequence analysis, with secondary structural considerations producing phylogenies that more closely reflect current ultrastructure-based modern taxonomy.47,48 This study supports that conclusion, demonstrating that secondary structure fold analysis offers an independent, structure-guided validation layer that enhances confidence in phylogenetic assignments.49,50
The SNP analysis disclosed significant genetic variation within species, notwithstanding the high similarity of 16S rRNA sequences, which exceeded 97% identity.51,52 The isolates of Staphylococcus hominis demonstrated relatively low SNP loads, ranging from 3-8 SNPs per isolate, with nucleotide divergence spanning 0.29%-0.70%. This observation is congruent with a close evolutionary grouping and minimal differentiation between strains. Remarkably, the S. hominis E3 isolate exhibited the highest SNP quantity, possessing 8 SNPs with a divergence of 0.70% among the Staphylococcus isolates. This finding indicates either a greater evolutionary divergence from the reference strain in the GenBank database or the possibility of sampling a genetically distinct subspecies or strain lineage of S. hominis. The observed accumulation of SNPs in some isolates, specifically Dyella marensis with 11 SNPs corresponding to a 0.99% divergence and Pseudomonas aeruginosa with 14 SNPs reflecting a 1.40% divergence, is likely indicative of the significant evolutionary distances between these recovered soil isolates and the established reference sequences in GenBank.53 This trend suggests that environmental isolates may belong to genetically divergent lineages that are understudied and underrepresented in reference databases. Alternatively, the heightened SNP counts could reflect ongoing microevolutionary diversification within soil populations of these taxa, where multiple competing strains sustain distinct genotypic profiles within the same isolate culture.54 Also, the InDel analysis demonstrated significant clustering of insertions within Pseudomonas aeruginosa W3, specifically between positions 20 and 50, encompassing 71% of InDels. This pattern suggests the presence of a localized mutational hotspot or an area of sequence instability.55 The confinement of all identified InDels to single-nucleotide occurrences (1 bp insertions/deletions), devoid of multi-nucleotide rearrangements, corroborates the hypothesis that 16S rRNA genes are under strong purifying selection against extensive deletions or insertions that could potentially disrupt ribosomal assembly.56
This investigation of culturable bacterial communities across the Makkah Region elucidates the complex interplay between physicochemical soil properties and microbial assemblage structure in arid terrestrial ecosystems. The Makkah Region exhibits pronounced spatial variability in critical soil parameters, particularly soil moisture (0.50-12.0%, 24-fold variation), reflecting geographic transitions from coastal plains to inland highlands that generate distinct environmental niches. Alkaline pH values (7.00-7.81, mean 7.30 ± 0.29) align with established pedogenic characteristics of arid soils, while sand-rich textures (74.8% average) reflect aeolian and coastal depositional processes. Culturable bacterial abundance demonstrated significant heterogeneity (6-20 isolates per governorate), with Fair Capital (20 isolates, 20%) and Jeddah (15 isolates, 15%) governorates collectively accounting for 35% of total isolates. Notably, Fair Capital’s unexpectedly high bacterial abundance despite minimal soil moisture demonstrates that anthropogenic enrichment and soil modification can substantially modulate microbial populations independent of natural moisture constraints, representing a significant decoupling of traditional moisture and diversity relationships in arid environments. Molecular analysis identified seven bacterial species: three closely related Staphylococcus hominis strains (Firmicutes) with exceptional sequence similarity (>99.5%) and four phylogenetically distinct Gram-negative taxa (Proteobacteria) with sequence identity values of 97.0%-99.8% relative to GenBank references. Neighbor-joining phylogenetic analysis with bootstrap support values exceeding 99% at major clade nodes validated sequence-based classifications. Notably, the isolation of Dyella marensis, a rarely reported gamma-proteobacterium, contributes to knowledge of microbial diversity in arid Arabian soils. Secondary structure analysis of 16S rRNA demonstrated complete congruence between structural architecture and primary sequence phylogenetics, providing independent validation of taxonomic assignments. SNP analysis revealed 3-14 polymorphisms per isolate with divergence rates of 0.29%-1.40%, while insertion-deletion analysis identified mutational hotspots, particularly in Pseudomonas aeruginosa (71% of InDels within a 30 nucleotide segment). Scanning electron microscopy revealed pronounced variation in soil particle morphology and biofilm colonization, with finer-textured soils supporting denser biofilm networks and coastal regions exhibiting extensive biofilm development. This multi-faceted approach, integrating cultivation-based isolation, molecular identification, phylogenetic inference, and secondary structure analysis, provides a robust framework for bacterial characterization, surpassing single methodological approaches. Future research should employ cultivation-independent metagenomic analyses, functional characterization of isolates, temporal sampling, and experimental manipulations to determine environmental thresholds governing bacterial community shifts. These findings advance understanding of microbial adaptation to polyextreme environmental stress conditions and provide baseline data for assessing responses of arid soil microbiomes to climatic and anthropogenic perturbations in the Arabian Peninsula.
ACKNOWLEDGMENTS
None.
FUNDING
None.
DATA AVAILABILITY
All datasets generated or analyzed during this study are included in the manuscript.
ETHICS STATEMENT
Not applicable.
- Iqbal S, Begum F, Nguchu BA, Claver UP, Shaw P. The invisible architects: microbial communities and their transformative role in soil health and global climate changes Environ Microbiome. 2025;20(1)36.
Crossref - Sacca ML, Caracciolo BA, Di Lenola M, Grenni P. Ecosystem Services Provided By Soil Microorganisms. In: Lukac M, Grenni P, Gamboni M, eds. Soil Biological Communities and Ecosystem Resilience. Sustainability in Plant and Crop Protection. Springer, Cham.
Crossref - Sharma SK, Ramesh A, Sharma MP, et al. Microbial Community Structure and Diversity as Indicators for Evaluating Soil Quality. In: Lichtfouse E, eds. Biodiversity, Biofuels, Agroforestry and Conservation Agriculture. Sustainable Agriculture Reviews, vol 5. Springer, Dordrecht.
Crossref - Semenov MV, Zhelezova AD, Ksenofontova NA, Ivanova EA, Nikitin DA, Semenov VM, Microbiological Indicators for Assessing the Effects of Agricultural Practices on Soil Health: A Review. Agronomy-Basel. 2025;15(2)335.
Crossref - Vasquez-Dean J, Maza F, Morel I, Pulgar R, Gonzalez M. Microbial communities from arid environments on a global scale. A systematic review. Biol Res. 2020;53(1):29.
Crossref - Torsvik V, Øvreås L. Microbial Diversity, Life Strategies, and Adaptation to Life in Extreme Soils. In: Dion P, Nautiyal CS, eds. Microbiology of Extreme Soils. Soil Biology, vol 13. Springer, Berlin, Heidelberg.
Crossref - Rahman NSNA, Hamid NWA, Nadarajah K. Effects of Abiotic Stress on Soil Microbiome. Int J Mol Sci. 2021;22(16):9036.
Crossref - Wani AK, Akhtar N, Sher F, Navarrete AA, Americo-Pinheiro JHP. Microbial adaptation to different environmental conditions: molecular perspective of evolved genetic and cellular systems. Arch Microbiol. 2022;204(2):144.
Crossref - Al-Awthan YS, Mir R, Alatawi FA, et al. Metagenome analysis identified novel microbial diversity of sandy soils surrounded by natural lakes and artificial water points in King Salman bin Abdulaziz Royal Natural Reserve, Saudi Arabia. Life. 2024;14(12):1692.
Crossref - Helmi NR. Exploring the diversity and antimicrobial potential of actinomycetes isolated from different environments in Saudi Arabia: a systematic review. Frontiers in Microbiology, 2025;16.
Crossref - El-Bastawisy MM. Developing urban environment of residential areas, Makkah city, Saudi Arabia, case study: Khansah residential area. In: Proceedings of the 14th Annual Meeting of the Saudi Umran Society: Urbanism in Makkah – Reality and Future. Makkah, Saudi Arabia: Saudi Umran Society; 2007:1-17
- ElKashouty M, Khan MYA, Alharbi K, Pande CB, Subyani AM, Tian FQ. Hydrogeology and Hydrogeochemistry of Saline Groundwater Seepage Zones in Wadi Bani Malik Basin, Jeddah, Saudi Arabia: Impacts on Soil and Water Resources. Water. 2023;15(19):3464.
Crossref - Alghamdi AG, Aly AA, Majrashi MA, Ibrahim HM. Impact of climate change on hydrochemical properties and quality of groundwater for domestic and irrigation purposes in arid environment: a case study of Al-Baha region, Saudi Arabia. Environ Earth Sci. 2023;82(1):39.
Crossref - Nunan N, Schmidt H, RaynaudX. The ecology of heterogeneity: soil bacterial communities and C dynamics Philos Trans R Soc Lond B Biol Sci. 2020;375(1798):20190249.
Crossref - Alotaibi MO, Sonbol HS, Alwakeel SS, et al. Microbial Diversity of Some Sabkha and Desert Sites in Saudi Arabia. Saudi J Biol Sci. 2020;27(10):2778-2789.
Crossref - Alharbi NM, Alharthi AK, Alsamadani A, Almihmadi RA, Alaidaroos BA. Screening and Characterization of Soil Microbes Producing Antimicrobial Compounds in Makkah Province, Saudi Arabia. Biosci Biotechnol Res Asia. 2021;18(4):719-732.
Crossref - Einarsson GG, Boutin S. Techniques: culture, identification and 16S rRNA gene sequencing. The Lung Microbiome (ERS Monography). 2019:18-34
Crossref - Patel AK, Singhania RR, Pandey A. Production of microbial polysaccharides. In: Rehm BHA, ed. Microbial Production of Biopolymers and Polymer Precursors: Applications and Perspectives. Hoboken, NJ: John Wiley & Sons; 2012:107-135.
Crossref - Regueira-Iglesias A, Balsa-Castro C, Blanco-Pintos T, Tomas I. Critical review of 16S rRNA gene sequencing workflow in microbiome studies: From primer selection to advanced data analysis. Mol Oral Microbiol. 2023;38(5):347-399.
Crossref - Lahlali R, Ibrahim DSS, Belabess Z, et al. High-throughput molecular technologies for unraveling the mystery of soil microbial community: challenges and future prospects. Heliyon. 2021; 7(10):e08142.
Crossref - Fathy WA, Techen N, Elsayed KNM, et al. Applying an internal transcribed spacer as a single molecular marker to differentiate between Tetraselmis and Chlorella species. Front Microbiol. 2023;14:1228869.
Crossref - Yasir M, Azhar EI, Khan I, et al. Composition of soil microbiome along elevation gradients in southwestern highlands of Saudi Arabia. BMC Microbiol. 2015;15:65.
Crossref - Al-Mutairi KA. Do Spatially Structured Soil Variables Influence the Plant Diversity in Tabuk Arid Region, Saudi Arabia? Sustainability. 2022;14(5):2611.
Crossref - Kim JM, Roh AS, Choi SC, et al. Y, Soil pH and electrical conductivity are key edaphic factors shaping bacterial communities of greenhouse soils in Korea. J Microbiol. 2016;54(12):838-845.
Crossref - Neilson JW, Califf K, Cardona C, et al. Significant Impacts of Increasing Aridity on the Arid Soil Microbiome. mSystems. 2017;2(3).
Crossref - Buelow HN, Winter AS, Van Horn DJ, et al. Microbial Community Responses to Increased Water and Organic Matter in the Arid Soils of the McMurdo Dry Valleys, Antarctica. Front Microbiol. 2016;7:1040.
Crossref - Gao Y, Xu X, Ding J, et al. The Responses to Long-Term Water Addition of Soil Bacterial, Archaeal, and Fungal Communities in A Desert Ecosystem. Microorganisms. 2021;9(5):981.
Crossref - Lopes AR, Becerra-Castro C, Vaz-Moreira I, Silva MEF, Nunes OC, Manaia CM. Irrigation with Treated Wastewater: Potential Impacts on Microbial Function and Diversity in Agricultural Soils. In: Fatta-Kassinos D, Dionysiou D, Kümmerer K, eds. Wastewater Reuse and Current Challenges. The Handbook of Environmental Chemistry, vol 44. Springer, Cham. 2015:105-128.
Crossref - Korbel KL, Stephenson S, Hose GC. Sediment size influences habitat selection and use by groundwater macrofauna and meiofauna. Aquat Sciences. 2019;81(2):39.
Crossref - Skariah S, Abdul-Majid S, Hay AG, et al. Soil Properties Correlate with Microbial Community Structure in Qatari Arid Soils. Microbiol Spectr. 2023;11(2):e0346222.
Crossref - Crits-Christoph A, Robinson CK, Barnum T, et al. Colonization patterns of soil microbial communities in the Atacama Desert. Microbiome. 2013;1(1):28.
Crossref - Gittings JA, Dall’Olmo G, Tang W, et al. An exceptional phytoplankton bloom in the southeast Madagascar Sea driven by African dust deposition. PNAS Nexus. 2024;3(10):386.
Crossref - Thomazini A, Francelino MR, Pereira AB, Schunemann AL, Mendonca ES, Schaefer CEGR. The spatial variability structure of soil attributes using a detailed sampling grid in a typical periglacial area of Maritime Antarctica. Environ Earth Sci. 2018;77(18):637.
Crossref - Pang H. Evaluation and modeling of risk factors associated with microbial contamination in produce pre-harvest environment [PhD thesis]. University of Maryland, College Park; 2017.
- Vásquez-Arroyo J, Vásquez-Arroyo J, Blanco-Contreras E, et al. Long-term impact of maize agroecological management on bacterial communities and soil health in the arid north of Mexico. Tropical and Subtropical Agroecosystems. 2023;26(3)
Crossref - Soussi A, Ferjani R, Marasco R, et al. Plant-associated microbiomes in arid lands: diversity, ecology and biotechnological potential. Plant Soil. 2016;405(1-2):357-370.
Crossref - Anand R. Endophytic colonization and nitrogen fixation by Paenibacillus polymyxa in association with lodgepole pine and western redcedar [PhD thesis]. University of British Columbia; 2010.
Crossref - Guaschino M, Garello M, Nari L, Spadaro D. Characterization of the soil, rhizosphere and root microbiome associated to kiwifruit vine decline syndrome in Italy. In: Proceedings of the 12th International Congress of Plant Pathology; August 20–25, 2023; Lyon, France. https://iris.unito.it/handle/2318/1947206
- Lee DW, Lee SD. Dyella marensis sp. nov, isolated from cliff soil. Int J Syst Evol Microbiol. 2009;59(6):1397-1400.
Crossref - Zhang L, Thomas JC, Miragaia M, et al. Multilocus Sequence Typing and Further Genetic Characterization of the Enigmatic Pathogen, Staphylococcus Hominis. Plos One. 2013;8(6):e66496.
Crossref - Calkins S, Couger MB, Jackson C, et al. Draft Genome Sequence of Staphylococcus Hominis Strain Hudgins Isolated From Human Skin Implicates Metabolic Versatility and Several Virulence Determinants. Genomics Data. 2016;10:91-96.
Crossref - Datta P, Banerjee S, Naha A, Pal S, Konar J, Sahu C. Staphylococcus Hominis Subsp. Hominis Causing Non – Lactational Breast Abscess: A Rare Pathogen Reported. J Evol Med Dental Sci. 2015;4(16):2830-2832.
Crossref - Muro MAd, Shuryak I, Tillman A, et al. The Abundance of the Potential Pathogen Staphylococcus Hominis in the Air Microbiome in a Dental Clinic and Its Susceptibility to Far-Uvc Light. Authorea. 2023.
Crossref - Ashimoto A, Hamada T, Adachi A, Tanigawa T, Tanaka Y. Molecular Epidemiology of Staphylococcus Spp. Contamination in the Ward Environment: Study on mecA and femA Genes in Methicillin-Resistant Strains. Kansenshogaku Zasshi. 1995;69(1):15-20.
Crossref - Hughes GL, Garay JAR, Koundal V, Rasgon JL, Mwangi MM. Genome Sequences of Staphylococcus hominis Strains ShAs1, ShAs2, and ShAs3, Isolated from the Asian Malaria Mosquito Anopheles stephensi. Genome Announc. 2016;4(2).
Crossref - Rivas E. Evolutionary conservation of RNA sequence and structure. Wiley Interdiscip Rev RNA. 2021;12(5):e1649.
Crossref - Rehakova K, Johansen JR, Bowen MB, Martin MP, Sheil CA. Variation in secondary structure of the 16S rRNA molecule in cyanobacteria with implications for phylogenetic analysis. Fottea. 2014;14(2):161-178.
Crossref - Wilmotte A, Herdman M. Phylogenetic relationships among the cyanobacteria based on 16S rRNA sequences, 978-0-387-98771-2. 2001 https://hdl.handle.net/2268/96343
- Chandra P, Chandra A. Elucidation of rRNA secondary structure and phylogenetic analysis of Salmonella enterica based on 16S RNA. Int J Curr Microbiol Appl Sci. 2017;6(7):4056-4063.
Crossref - Lavender CA, Lorenz R, Zhang G, Tamayo R, Hofacker IL, Weeks KM. Model-Free RNA Sequence and Structure Alignment Informed by SHAPE Probing Reveals a Conserved Alternate Secondary Structure for 16S rRNA. PLoS Comput Biol. 2015;11(5):e1004126.
Crossref - Johnson JS, Spakowicz DJ, Hong BY, et al. Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat Commun. 2019;10(1):5029.
Crossref - Maretto F, Reffo E, Dalvit C, Barcaccia G, Mantovani R. Finding 16S rRNA gene-based SNPs for the genetic traceability of commercial species belonging to Gadiformes. Ital J Anim Sci. 2007;6(sup1):161-163.
Crossref - Mohkam M, Nezafat N, Berenjian A, Mobasher MA, Ghasemi Y. Identification of Bacillus Probiotics Isolated from Soil Rhizosphere Using 16S rRNA, recA, rpoB Gene Sequencing and RAPD-PCR. Probiotics Antimicro Prot. 2016;8(1)8-18.
Crossref - Li Y, Pinto-Tomas AA, Rong X, Cheng K, Liu M, Huang Y. Population Genomics Insights into Adaptive Evolution and Ecological Differentiation in Streptomycetes. Appl Environ Microbiol. 2019;85(7):e02555-02518.
Crossref - Darmon E, Leach DR. Bacterial genome instability. Microbiol Mol Biol Rev 2014;78(1):1-39.
Crossref - Maslunka C, Gifford B, Tucci J, Gurtler V, Seviour RJ. Insertions or deletions (Indels) in the rrn 16S-23S rRNA gene internal transcribed spacer region (ITS) compromise the typing and identification of strains within the Acinetobacter calcoaceticus-baumannii (Acb) complex and closely related members. PLoS One. 2014;9(8):e105390.
Crossref
© The Author(s) 2026. Open Access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License which permits unrestricted use, sharing, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.