


ISSN: 0973-7510
E-ISSN: 2581-690X
The etiological agent of coronavirus disease (COVID-19) that emerged at the end of year 2019 was first reported in Wuhan, China and was found to be SARS-CoV-2 (severe acute respiratory syndrome coronavirus-2). The massive COVID-19 waves were due to various variants. As per the reports of other study it was also found that Omicron variant spread faster than various other variant such as delta variant. Omicron has been reported from various countries and now from many states of India too. Therefore, keeping this in mind, this study was undertaken to study all the lineages of SARS-CoV-2 Omicron variant of disease COVID-19 that are circulating in the population of Uttarakhand with objective to study next generation sequencing of all the RT-PCR positive of SARS-CoV-2 and to find out all the lineages of the Omicron variant of SARS-CoV-2. This was a retrospective study conducted from 1st January 2022 to 30th September 2022. Next generation sequencing was performed on all the samples that were tested for COVID-19 by using Ion AmpliSeq kit on Ion Chef instrument. A total of 2149 samples were tested in which majority of samples belong to age group of 21-40 years. Males were affected more than females. BA.2 was found to be the predominant lineage of total of 46 lineages that were identified. Their mutations were also studied. We conclude that different variants of clade 21L, 22B, 22D and Omicron subvariant BA.2, BA.2.38 and BA.2.75 were the ones that were circulating amongst the population of Uttarakhand. The characteristic mutation that was found were T19I and V213G in NTD, S373P, S375F, T376A, and D405N in RBD.
SARS-CoV-2, Omicron, Lineages, Mutations, Genome Sequencing, Molecular Epidemiology
At the end of January 2020, WHO announced COVID-19 pandemic which is due to severe acute respiratory syndrome coronavirus -2 (SARS-CoV-2) as a public health emergency of international concern (PHEIC).1 Since then, it has been spreading all over the world in an impressing high rate. To combat infection and severity of disease, many vaccines were developed over time which showed their efficacy positively. COVID-19 being a viral etiology has capability to change its genetic makeup and undergo genetic evolution which confer major impact on its host range, re-infection, diagnosis and also its performance against vaccines and therapeutics.2 Because of this genetic evolution there have been emergence of new strain from the original wild type strain of SARS-CoV-2. Amongst the new variants that have emerged, four variants, namely Alpha (B.1.1.7), Beta (B.1.351), Gamma (P.1) and Delta (B.1.617.2) have been reported until late 2021. These variants not only made the pandemic period more crucial owing to their transmissibility, also contributed to the surge in cases all over the world.3 The current variant that has emerged is known by the name of Omicron variant of SARS-CoV-2 (B.1.1.529) and was first reported from South Africa on 14th November 2021 and soon circulated all over the world after which it was declared as variants of Concern (VOCs) by WHO as soon it was reported. The variant was thought to be different from Delta variant in its transmissibility and re-infection owing to increased neutralizing antibody escape mechanisms. This variant had several mutations in its structure such as in spike protein along with envelop protein, nucleocapsid protein and in open reading frame 1ab (ORF 1ab).4 The relative effective reproduction number of Omicron variant over Delta variant is estimated to be 3.19 times mainly because of these genetic changes. Approximately 18261 mutations are present in the genome of Omicron variant out of which coding region and extragenic regions constitute 97% and 558 mutations respectively. Receptor binding domain (RBD) of spike protein comprised of thirty mutations along with three other deletion and one insertion mutation present outside the spike protein.5 Mutations were also present in non-structural protein (NSP) part of Omicron and to name a few are: NSP3-K38R, V1069I, TIANET, A1892T, etc. and this NSPs of CoVs are essential for replication and transcription of virus. Therefore, compared to other variant of concern (VOCs) and early wild-type strain, Omicron variant exhibited largest number of mutations. As analyzed by GISAID (Global Initiative on Sharing All Influenza Data), N-terminus domain comprised of 11 mutations in preliminary data which included six deletions and one insertion with unique mutations such as N211 and ins214EPE.6
Absorbingly some mutations present in the RBD of Omicron variant are shared by Delta variants (two out of three) such as substitution of asparagine from lysine at 417 position and threonine to lysine substitution at position 478. Another mutation that is present in delta variant is leucine to arginine substitution at 452 position which is not present in Omicron variant and this is known to boost the affinity for ACE2 receptor that is found on the surface of human lungs and other body cells.7
Also, another important role in Omicron variant spread was played by waning immunity, that is, decrease in immunity induced by vaccines over time. Owing to the current knowledge about the mutations that are present in this variant, this study was undertaken to study more about the lineages of SARS-CoV-2 Omicron variant and which variants were prevalent in the state of Uttarakhand.
Study population
Upper respiratory tract samples from all age groups were included in this retrospective study that were received for SARS-CoV-2 diagnosis from January 2022 till September 2022, in Department of Microbiology, Govt. Doon Medical College, Dehradun. It included all the samples from in-patient, out-patients and community surveillance. This study was approved by the Institutional Ethics Committee, GDMC, Dehradun.
Study samples
Samples were collected in 3ml viral transport media tube according to standard protocol for sequencing of the samples that were tested positive. The samples for sequencing obtained from various other hospitals were received in cryovials after they were tested positive for SARS-CoV-2.
RNA isolation
Biosafety level 2 (BSL) facility was used for RNA extraction using QIAsymphony RNA kit (Qiagen, Hilden, Germany) and the method employed for the same was automated magnetic bead-based extraction method. QIA symphony instrument (Qiagen, Hilden, Germany) was used for extraction as instructed by the manufacturer in which 200 μl of viral transport media (VTM) was transferred to a 96-well deep well cartridge plate that is supplied with the kit.
Library preparation and sequencing
Ion AmpliSeqTM Library kit Plus (Thermo Fisher Scientific, Waltham, Massachusetts, United States) were used for library preparation according to manufacturer’s instruction. First the samples were isolated and quantification of viral RNA was done. For Ion AmpliSeqTM SARS-CoV-2 Research panel library (Thermo Fisher Scientific, Waltham, Massachusetts, United States), sample containing as little as 20 copies of viral RNA (10 copies per target amplification reaction) was used. This research panel consists of two 5X primer pair pools that targets 23 amplicon which are specific to the SARS-CoV-2 coronavirus, and 5 human expression controls. The panel with amplicon length of range 125-25 bp provides almost 99% coverage of SARS-CoV-2 genome and covers all potential serotypes. MagMAXTM Viral/Patogen Nucleic Acid Isolation Kit (Thermo Fisher Scientific, Waltham, Massachusetts, United States) was used for RNA isolation. TaqManTM 2019-nCoV Assay Kit v1, TaqManTM 2019-nCoV Control Kit v1, and TaqPathTM 1-step RT-qPCR master Mix (Thermo Fisher Scientific, Waltham, Massachusetts, United States) was used for quantification and normalization of amount of RNA. Ct result was used to estimate copy number in sample for each 2019-nCoV assay. cDNA synthesis from each isolate was done using SuperScriptTM VILOTM Mastermix (Thermo Fisher Scientific, Waltham, Massachusetts, United States) as per user manual. Amplification of the target regions was done with two primer pools protocol and after that target amplification reaction were combined. Then, partial digestion of amplicon is carried out using FuPa Reagent. Different barcode adaptors were added to each library by using Ion XpressTM Barcode Adapters 1-96 kit (Thermo Fisher Scientific, Waltham, Massachusetts, United States) according to manufacturer’s instruction in which IonCodeTM Adapters (Thermo Fisher Scientific, Waltham, Massachusetts, United States) including both forward and reverse adapters in single well are provided at optimum concentration. After addition of these barcodes, purification of the library is done by using Agencourt™ AMPure™ XP Reagent (Beckman Coulter Life Sciences, Indianapolis, United States). QubitTM Fluorometer (Thermo Fisher Scientific, Waltham, Massachusetts, United States) or AgilentTM 2100 BioanalyzerTM instrument (Agilent, Santa Clara, United States) was used for the quantification of amplified library. The libraries that did not meet the targeted concentration were reamplified and reassessed by fluorometry (QubitTM dsDNA HS Assay kit). These libraries were then loaded in the chip and these chips were subjected to templating which was performed by the Ion ChefTM Instrument in which all the reagents and solution cartridges are loaded previously. After that next generation sequencing is performed on the Ion GeneStudioTM S5 System with the use of Ion 540TM Kit-Chef and Ion 540TM Chip Kit (Thermo Fisher Scientific, Waltham, Massachusetts, United States). Analysis of the sequenced results were done in Torrent suiteTM Software (Thermo Fisher Scientific, Waltham, Massachusetts, United States) with SARS-CoV-2 plugins: COVID-19 Annotate Snpeff, IRMA report, and the Assembler Trinity plugins to perform analysis with standard configuration. For lineage assignment by Pangolin COVID-19 Lineage Assigner, FASTA files of sequences were used that have come from libraries with optimal concentration. These FASTA files are text-based format for representing DNA sequences using single-letter code. These FASTA files were then submitted to IBDC (Indian Biological Data Centre) according to INSACOG data submission guidelines for analysis.
Data analysis
The data of the patients were retrieved from the excel sheet prepared after the final result of sequencing was obtained. Basic demographic data was taken from the excel sheet.
A total of 2149 samples were tested during the study period. The mean age of presentation was 37.9±18.45 years and majority of patients were in the age group of 21-40 years. Male patients were affected more than female patients.
Table 2 shows the predominant lineages in which BA.2 was reported in majority of the samples which was followed by BA.2.75. Apart from these BA.2.38, BA.2.5.1, BA.2.75.2, BA.2.76 and BA.5.2 were also present.
The major mutations that were present in all lineages were G339D, S373P, S375F, K417N, N440K, G446S, S477N, T478K, E484A, Q493R, Q498R, N501Y, Y505H. These mutations were present in spike protein at receptor binding domain locations. These amino acid substitutions are also described by CDC.8 In other structural gene, E protein contains T9I mutations, M protein contains Q19E and A63T mutation and N protein contains P13L, R203K, G204R and S413R mutations. N gene in different lineages of Omicron also contains deletion of 31-33 amino acid position. Rest of the genome analysis showed that in ORF1a gene, in all the variants, common mutations were present such as S135R, L3027F, T3090I, P314L, R1315C, T2163I and L3201F. However in lineages such as BA.2, BA.2.38 and BA.5.2, BA.2.75, one deletion in the gene del3675/3677 was also present and its sub-lineages also comprised of additional mutations such as S1221L, P1640S, N4060S and G662S. These mutations being exclusive for this lineage. In all detected lineages of Omicron, ORF6 showed D61L mutation except BA.5.2, while ORF8 showed mutation S84L.
Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) was declared pandemic by WHO in march 2020 soon after its emergence in Wuhan, China in December 2019 due to its rapid spread in the whole world. After its emergence, different waves were observed in many parts of the world due to the mutations in the genome of COVID-19. These waves were caused by different variants and their lineages and these lineages differ from each other in terms of their transmission potential and immune escape potential. The number of COVID-19 cases is on continuous rising trend all across the world.
India has observed three different waves of COVID-19 since its emergence. The different variant that was responsible for these waves were known by the names of alpha, beta, gamma, delta and so on. These variants were identified with the help of whole genome sequencing (WGS) which serves an important tool for various reasons. Firstly, it is helpful for the study of virus evolution over time and also predicting the trends of disease transmission. Also, it helps to determine the geographic prevalence of disease. Lastly, it can help understand the most effective design and platform for developing vaccines and therapeutics.
The second wave of coronavirus disease 2019 was due to delta variant that emerged in April 2021.9 This variant was responsible for high morbidity and mortality as well as increase in number of hospitalization, high requirement of oxygen and cases of mucormycosis too. These were the reasons why sequencing the genome of coronavirus became important to know the variant responsible for high morbidity and mortality.
Omicron variant of COVID-19 emerged in month of November 2021 in South Africa and spread to other parts of the world. This study was conducted to reveal the most predominant lineage of Omicron variant that emerged as third wave of COVID-19 in January 2022 in different districts of Uttarakhand.
In the present study, a total of 2149 samples were studied during a period of 09 months (January 2022 till September 2022). These samples were positive for COVID-19 and were received from different districts of Uttarakhand. Demographic data was collected for all the samples and mean age of presentation of the patient included in the study was 37.9±18.45 years and most common age group to be affected was 21-40 years (45.3%) followed by 41-60 years (24.8%) as shown in Table 1. This finding of our study was consistent with the study conducted by Raju et al. in Chennai, Garg et al. in Delhi and Zaman et al. in Gorakhpur with the mean age of 37 years, 3 years and 33 years respectively.10,11,12 Male preponderance over females was also a finding in our study which also corroborated with the findings of the study mentioned above.
Table (1):
Demographic data of all the samples tested
| Parameter | N (%) |
|---|---|
| Total Sample | 2149 (100) |
| Age (mean±SD) | 37.9±18.45 |
| Age (years) | |
| 0-20 | 342 (15.9) |
| 21-40 | 975 (45.3) |
| 41-60 | 534 (24.8) |
| 61-80 | 265 (12.3) |
| >80 | 33 (1.5) |
| Gender | |
| Male | 1167 (54.3) |
| Female | 982 (45.6) |
| Vaccination status | |
| Yes | 528 (24.5) |
| Unknown | 1621 (75.4) |
| Type of vaccine (among vaccinated) | |
| Covaxin | 48 (9.0) |
| Covishield | 469 (88.8) |
| Jcovden | 1 (0.18) |
| Comirnaty | 9 (1.7) |
| unknown | 1 (0.18) |
| Hospitalization status (amongst vaccinated) | |
| Yes | 152 (28.7) |
| No | 376 (71.2) |
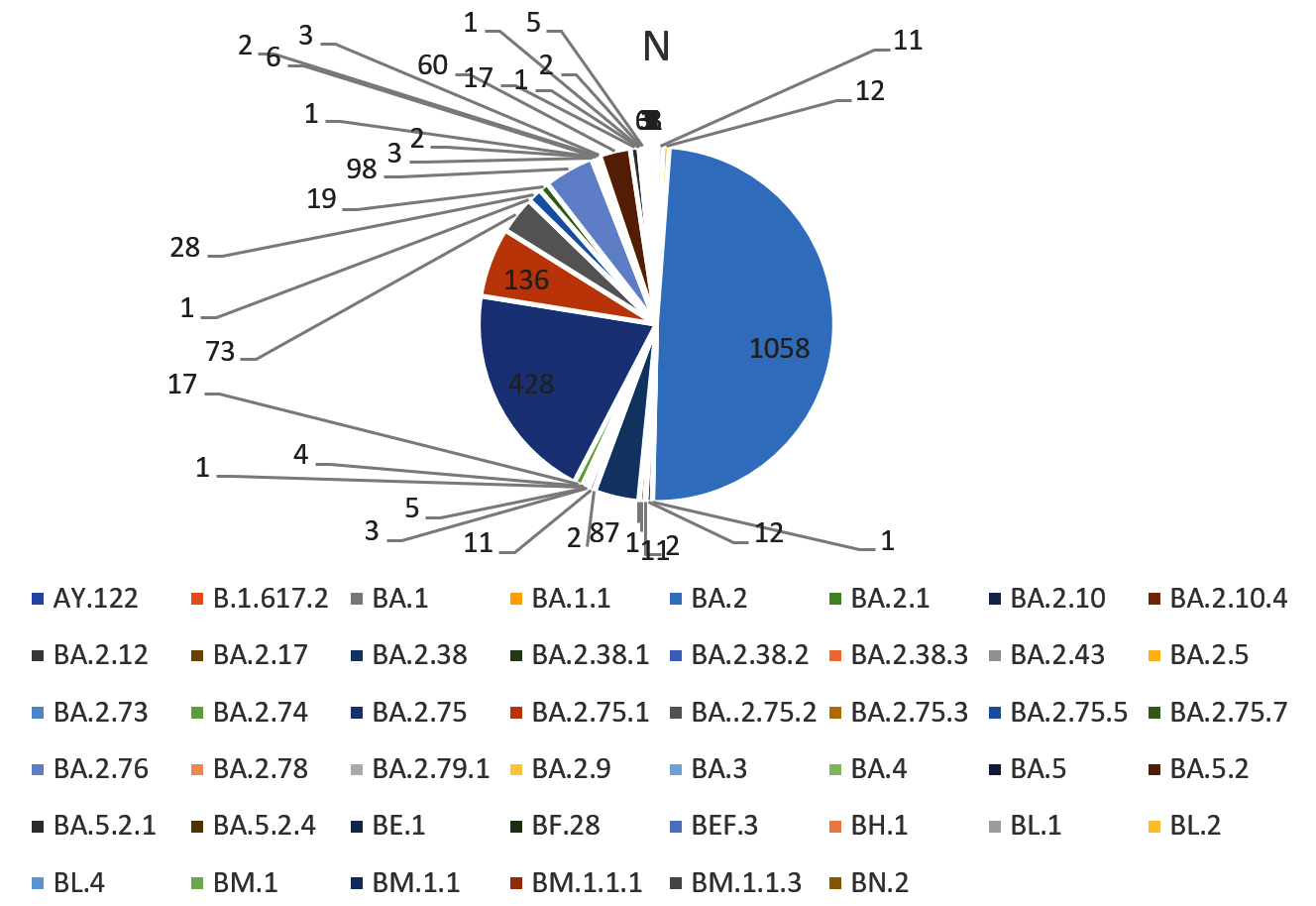
Figure 1. All the lineages present in the population of Uttarakhand
In the present study, sequencing of the samples from January 2022 showed that majority of the samples belonged to Omicron variant and only two samples were positive for delta variant which suggested that the peak in the third wave of COVID-19 was due to Omicron variant and delta variant (predominant in second wave) was replaced by Omicron variant. Figure 1 shows all the lineages that were detected in the samples sequenced. Table 2 shows the predominant lineages of omicron variant in which BA.2 was detected in the majority of the samples followed by BA.2.75. Delta variant was predominant till December 2021 and this finding was also consistent with the findings of the study conducted by Sharma et al. in Rajasthan who performed WGS analysis of SARS-CoV-2 positive cases from Nov. 2021 to Jan. 2022.13 Potdar et al. from Indian Council of Medical Research, National Institute of Virology, Pune also studied the effect of Omicron on Indian population after its emergence in South Africa and found that B.1.1.529 was responsible for infection in the initial cases whereas BA.1 lineage was responsible for infection in later contacts which also led to third wave of COVID-19.14 In more than 50 countries including United Kingdom, the United States, France, Italy, Netherlands, and India, Omicron variant caused new wave and this was reported in the study done by Ranjan in the year 2022.15
Table (2):
Incidence of predominant lineages that were circulating in population in total sample sequenced
Lineage |
Samples [N (%)] |
|---|---|
BA.2 |
1058 (49) |
BA.2.38 |
87 (4) |
BA.2.75 |
428 (19) |
BA.2.75.1 |
136 (6) |
BA.2.75.2 |
73 (3) |
BA.2.76 |
98 (4) |
BA.5.2 |
60 (2) |
In the present study, BA.1 lineage of Omicron was first detected on 3rd January 2022 along with some samples positive for BA.2 detected simultaneously. This sub-lineage (BA.1) of Omicron variant was considered as original form of Omicron variant as described by Rana et al.7 The findings of the present study depict that sub-lineage BA.2 started peaking from January 2022 soon after it was identified as shown in Table 3. This variant was also replaced by other subvariant of Omicron BA.2.38 which was first detected in May 2022 and peaked in June 2022 as shown in Figure 2. INSACOG (Indian SARS-CoV-2 Genomic Consortium), a national agency also reported that BA.2 Omicron variant was replaced with BA.2.38 as seen in this study.16
Table (3):
Trends in evolution of variant of concern from January 2022 till September 2022
Month/ Lineage |
BA.2 [N (%)] |
BA.2.38 [N (%)] |
BA.2.75 [N (%)] |
BA.2.75.1 [N (%)] |
BA.2.75.2 [N (%)] |
BA.2.76 [N (%)] |
BA.5.2 [N (%)] |
|---|---|---|---|---|---|---|---|
JANUARY |
394 (18.3) |
0 (0) |
0 (0) |
0 (0) |
0 (0) |
0 (0) |
0 (0) |
FEBRUARY |
380 (17.6) |
0 (0) |
0 (0) |
0 (0) |
0 (0) |
0 (0) |
0 (0) |
MARCH |
124 (5.7) |
0 (0) |
0 (0) |
0 (0) |
0 (0) |
0 (0) |
0 (0) |
APRIL |
50 (2.3) |
0 (0) |
0 (0) |
0 (0) |
0 (0) |
0 (0) |
0 (0) |
MAY |
13 (0.6) |
2 (0.09) |
0 (0) |
0 (0) |
0 (0) |
0 (0) |
0 (0) |
JUNE |
23 (1.0) |
44 (2.0) |
13 (0.6) |
1 (0.04) |
0 (0) |
15 (0.7) |
1 (0.04) |
JULY |
36 (1.6) |
36 (1.6) |
200 (9.3) |
39 (1.8) |
0 (0) |
55 (2.5) |
17 (0.8) |
AUGUST |
4 (0.18) |
5 (0.2) |
169 (7.8) |
64 (2.9) |
32 (1.4) |
17 (0.8) |
21 (0.9) |
SEPTEMBER |
1 (0.04) |
0 (0) |
33 (1.5) |
30 (1.4) |
32 (1.4) |
10 (0.4) |
18 (0.8) |
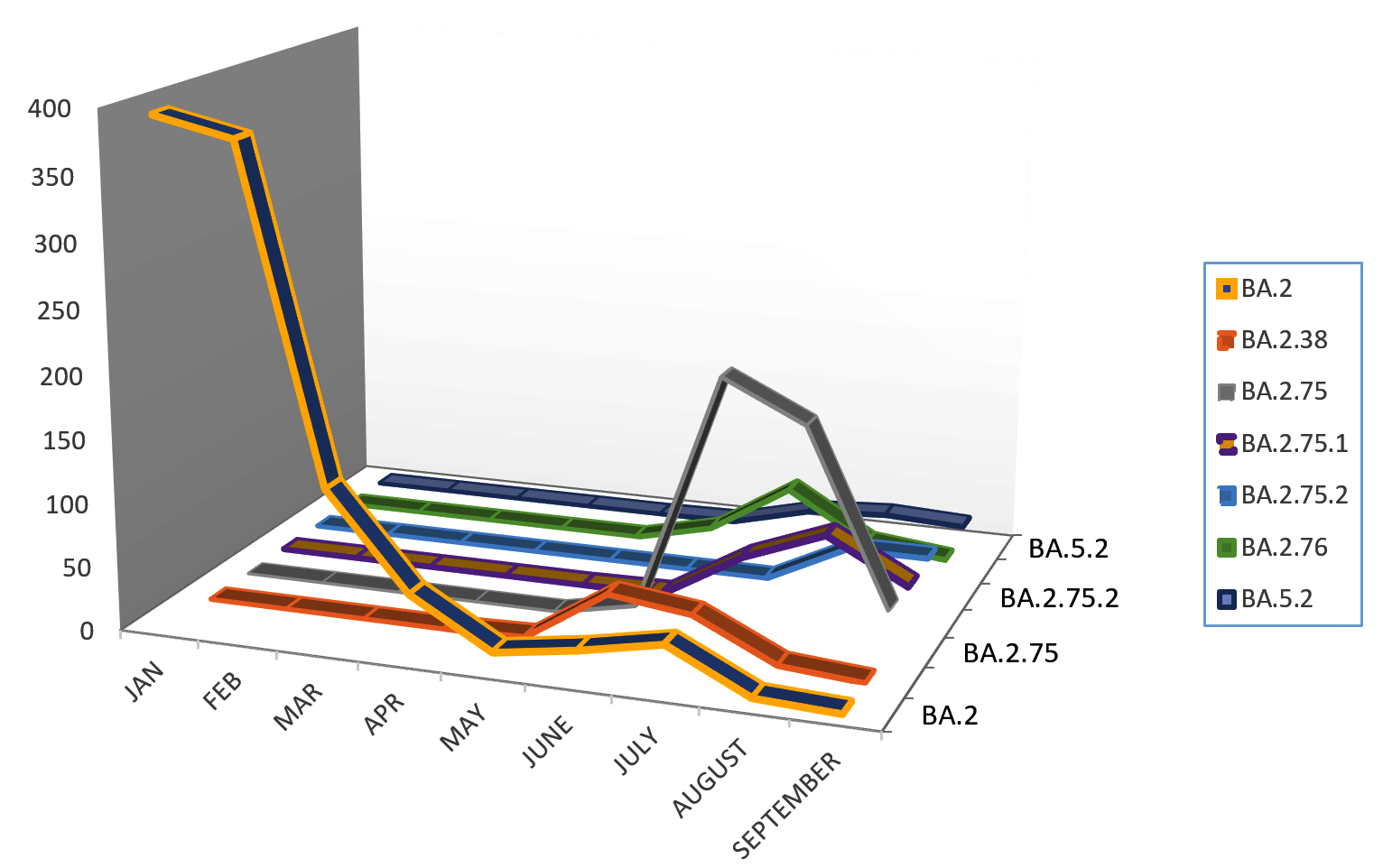
Figure 2. Trends in evolution of variant of concern from January 2022 till September 2022
However, this variant was also soon replaced by BA.2.75 that was first identified in the month of June 2022 but peaked in July 2022. BA.2.75, BA.2.12.1, BA.4 and BA.5 were also placed as Variant of Concern Lineage Under Monitoring (VOC-LUM) in July 2022 according to WHO.17 In India, at least 23 samples of BA.2.75 were recorded at the time of its emergence in the data uploaded on Nextstrain which is an open-source platform of genomic data and these were reported from areas such as Maharashtra, Karnataka, and Jammu & Kashmir. This variant was also detected outside India, such as in Australia, Canada, Germany and New Zealand.18 In the present study also, BA.2.75 was mainly reported near the month of July and August 2022 which makes it a consistent finding. Tamil Nadu and Telangana were those southern states from where BA.5 was mainly reported. In the present study, various other mutations were also identified from NGS such as BA.2.75.1, BA.2.75.2, BA.2.76 and BA.5.2, however, the most predominant lineage of them all was BA.2.75 which was present in maximum number of samples as shown in Figure 2.
In the present study, amongst the most predominant lineages that were circulating in Uttarakhand, we observed that in Dehradun district majority of the cases were of BA.2 lineage which was replaced by its sub-lineages BA.2.75 and BA.2.75.1 in the month of July, whereas, in Haridwar district the majority of the cases were of BA.2 lineage followed by BA.2.75 lineage which was present in few cases. Other districts majorly showed the presence of BA.2 lineage as shown in Figure 3 and Figure 4. There were some other lineages also that were circulating in the population of Uttarakhand such as BA.2.76, BA.2.75.1, BA.2.75.2 and BA.5. These all lineages were reported from all districts of Uttarakhand with majority being reported from Dehradun and Haridwar. This observation was between the period of 9 months (Jan. 2022 till Sept. 2022). Similarly, in a study done in Delhi, initial few cases were of BA.1 lineage (73.1%) while BA.2 lineage was present in 26.8% cases amongst all the cases in December 2021. Zaman et al. from Gorakhpur also found a shift of sub-lineages of Omicron variant in their study where amongst the samples of January and February 2022 the majority were of BA.2 sub-lineage (82.3%).12
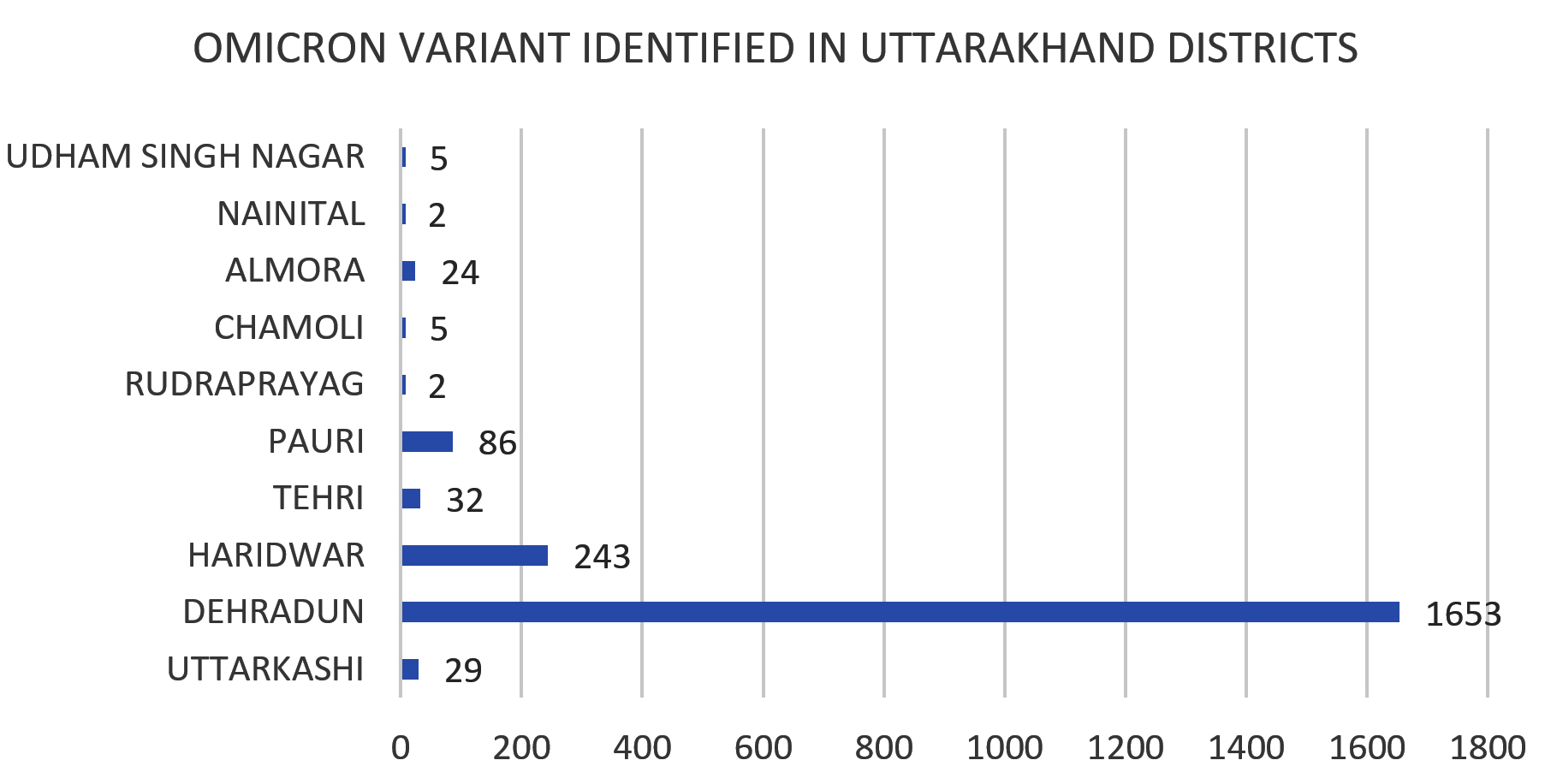
Figure 3. Distribution of Omicron variant in different districts of Uttarakhand
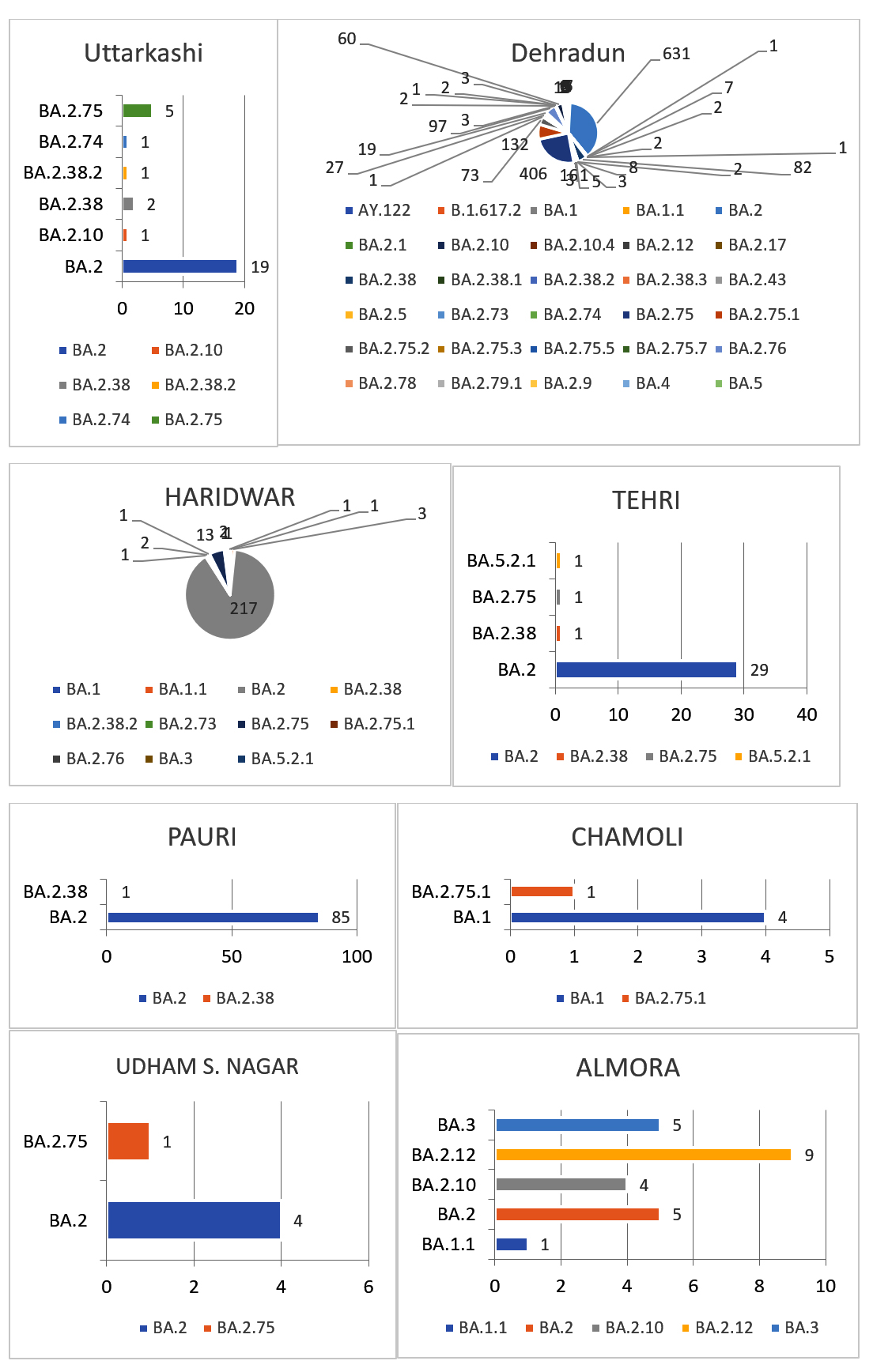
Figure 4. Different lineages present in different districts of Uttarakhand
The presence of this high number of Omicron lineage and sub-variant in the population of Uttarakhand and this shift of lineages from one to other at different point of time suggests that there was persistence of Omicron variant in the population and the virus kept on mutating to survive in the population. The presence of these mutant strain can also be due to the fact that Uttarakhand being high in tourism, so the people coming from other states can also bring the mutant strain from outside. It was also noticed that the peaks of these mutant strain corresponded to the month that were high in tourism since no major lockdown was imposed in the state so there was free interstate transmission of people.
In the present study, there were several mutations that were identified by Next Generation Sequencing in the structure of Omicron variants that confers various benefits to the virus for its maintenance and survival in the population as shown in Table 4. These mutations are responsible for immune escape, increasing binding capacity, improves cell surface entrance and effective cellular invasion. However, due to the fact that vaccination was given to mass population worldwide and also due to the active immunity acquired during infection, the severity of the disease was not much increased in spite of these high number of alterations in the Omicron variant.19 Figure 5 shows the phylogenetic distribution of SARS-CoV-2 genome in the population of Uttarakhand where all the genome was identified by next generation sequencing. However, phylogenetic tree of the predominant lineage that was identified (BA.2.75 and BA.2) was also constructed through INSACOG portal which is depicted in Figure 6a and 6b. The major mutations that were present in all the lineages were G339D, S373P, S375F, K417N, S477N, Q493R, N501Y, N440K, G446S, T478K, E484A, Q498R, Y505H. The location of these mutations was in the spike protein (receptor binding domain). These amino acid substitutions are also described by CDC.8 In other structural gene, E protein contains T9I mutation, M protein contains Q19E and A63T mutation and N protein contains P13L, R203K, G204R and S413R mutations. N gene in different lineages of Omicron also contains deletion of 31-33 amino acid position. Rest of the genome analysis showed that in ORF1a gene, in all the variants, common mutations were present such as S135R, L3027F, T3090I, P314L, R1315C, T2163I and L3201F. However, in lineages such as BA.2, BA.2.38 and BA.5.2, BA.2.75, one deletion in the gene del3675/3677 was also present and its sub-lineages also comprised of additional mutations such as S1221L, P1640S, N4060S and G662S. These mutations being exclusive for this lineage. In all detected lineages of Omicron, ORF6 showed D61L mutation except BA.5.2, while ORF8 showed mutation S84L.
Table (4):
Lineages that were identified in the study, including their mutations detected by NGS
| Variant | Lineage | Mutations | ||
|---|---|---|---|---|
| ORF1a | S | N | ||
| OMICRON | BA.2 | S135R,T842I,G1307S,L3027F,P3395H,R1315C,P314L,T2163I,L3201F,DEL3675/3677 | T19I,V213G,S31F,T376A,D405N,N440K,E484A,Y505H,D614G,N679K,P681H,D796Y,Q954H,N969K,L24S,del25/27,G142D,G339D,K417N,Q493R | P13L,R203K,G204R,S413R,del31/33 |
| OMICRON | BA.2.38 | S135R,T842I,G1307S,L3027F,T2163I,L3201F,DEL3675/3677 | T19I,V213G,S371F,T376A,D405N,N440K,E484A,Y505H,D614G,N679K,P681H,D796Y,Q954H,N969K,L24S,del25/27,G142D,G339D,K417N,Q493R | P13L,R203K,G204R,S413R,del31/33 |
| OMICRON | BA.2.75 | S135R,T842I,G1307S,L3027F,T2163I,L3201F,S1221L,P1640S,N4060S,G662S | T19I,V213G,S371F,T376A,D405N,N440K,E484A,Y505H,D614G,N679K,P681H,D796Y,Q954H,N969K,G257S,G339H,G446S,N460K | P13L,R203K,G204R,S413R |
| OMICRON | BA.2.75.1 | S135R, T842I, G1307S, L3027F, T2163I, L3201F, S1221L, P1640S, N4060S, G662S | T19I, V213G, S371F, T376A, D405N, N440K, E484A, Y505H,D614G,N679K,P681H,D796Y,Q954H,N969K,L24S,del25/27,G142D,G339D,K417N,Q493R | P13L, R203K, G204R, S413R, del31/33 |
| OMICRON | BA.2.75.2 | S135R, T842I, G1307S, L3027F, T2163I, L3201F, S1221L, P1640S, N4060S, G662S | T19I,V213G,S371F,T376A,D405N,N440K,E484A,Y505H,D614G,N679K,P681H,D796Y,Q954H,N969K,G257S,G339H,G446S,N460K | P13L, R203K, G204R, S413R |
| OMICRON | BA.2.76 | S135R,T842I,G1307S,L3027F,T2163I,L3201F,DEL3675/3677 | T19I,V213G,S371F,T376A,D405N,N440K,E484A,Y505H,D614G,N679K,P681H,D796Y,Q954H,N969K,L24S,del25/27,G142D,G339D,K417N,Q493R | P13L, R203K, G204R, S413R, del31/33 |
| OMICRON | BA.5.2 | S135R, T842I, G1307S, L3027F, T2163I, DEL 3675/3677, T1050N | T19I,V213G,S371F,T376A,D405N,N440K,E484A,Y505H,D614G,N679K,P681H,D796Y,Q954H,N969K,L24S,del25/27,G142D,G339D,K417N,del69/70,L452R,F486V | P13L, R203K, G204R, S413R, del31/33 |
Note: red and bold indicates the major mutations present in different lineages of Omicron variant in ORF1a, S and N gene.
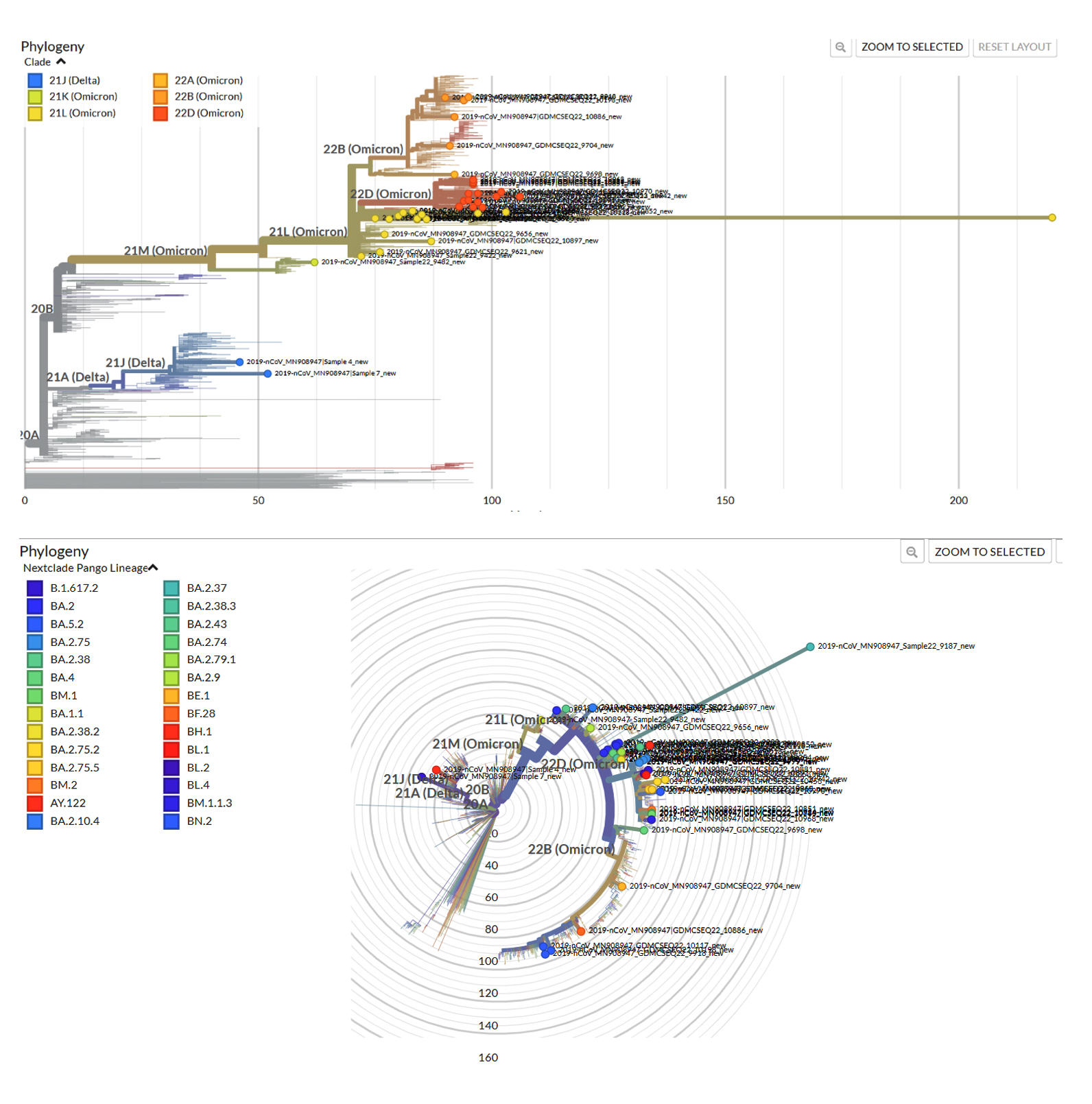
Figure 5. Phylogenetic distribution of SARS-CoV-2 genome in Uttarakhand
(phylogenetic tree of Omicron built using the Nextclade online tool)
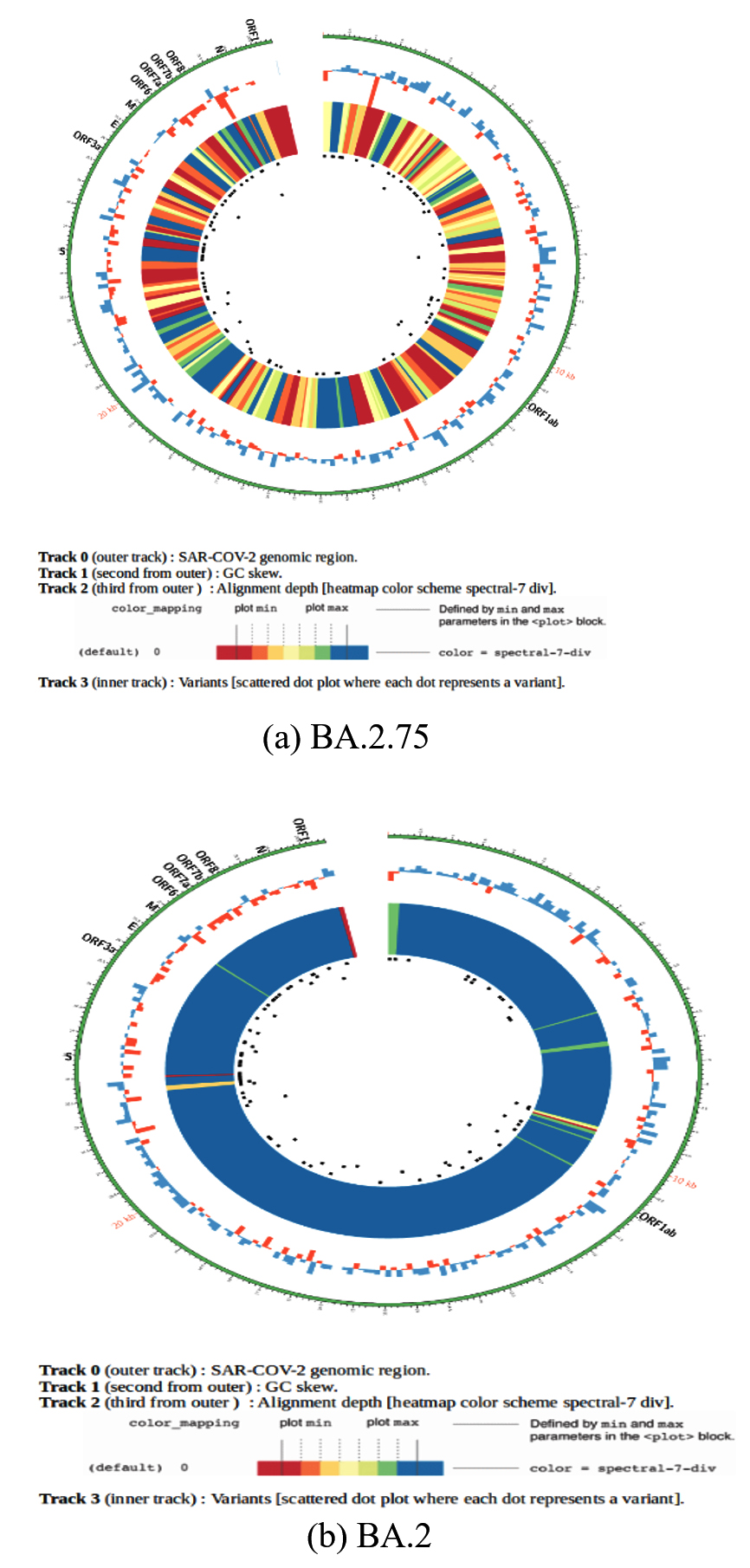
Figure 6. Phylogenetic tree of predominant lineages: (a). BA.2.75; (b). BA.2
(Picture recovered from INSACOG IBD Platform)
In the lineage BA.2 which was the most predominant lineage as identified by NGS in our study, the mutations that were identified were responsible for the increased affinity of S-protein towards the ACE2 receptors and also lead to increased transmissibility of the Omicron variant.20 Mutations such as N764K, D796Y, Q954H and N969K have been responsible for change in entry of virus in the host cell and also transmissibility.21
Mutations such as P681H, H655Y, and N679K present in the variants are associated with increased pathogenicity by facilitating S-protein cleavage by furin and thereby helping in transmission.18 Increase in transmissibility and infectiousness of the disease is also associated with several other mutations such as Q498R, S477N, and N501Y which are also responsible for increase in binding capacity of S-protein with ACE2 receptors. However, mutations such as T478K and E484A are those that are responsible for increase in neutralization of antibody resistance and higher immune escape.22,23 Additionally, the infectivity of SARS-CoV-2 virus was increased due to the D614G substitution which is located in the S protein and is conserved in all the variant of Omicron.24 The mutations present in nucleocapsid protein (R203K and G204R) are linked with enhanced sub genomic RNA expression and viral replication.25
The sub-variant BA.2.75 soon displaced other sub-lineages due to its adaptation to new mutations. This variant also showed large number of mutations in the viral spike protein amongst which some were similar to the mutations that were present in the other lineages but some mutations were different such as G257S, D339H, N460K, and G446S. Higher infectivity of BA.2.75 (up to 44-fold) was due to the mutation such as Q493R, N460K and alteration of G339D to G339H which in all together impose fusogenicity.26 The mutation G446Shave potential to affect immune evasion and ACE2 binding. It can lead to considerable immunological escape which suggest that reinfection and breakthrough infections may be leading to the spread of BA.2.75 and thus increasing the number of cases.27,28 Immune evasion was reported more in BA.2.75 lineage than BA.5 lineage especially in cases which were infected earlier with delta variant.29 This fast transmission of BA.2.75 could be due to the mutation such as S1221L, P1640S present in ORF1a gene which was absent in BA.2.
Limitation
The present study’s limitation include that no data was available about the clinical status of the patient due to which a potential relationship between the predominant lineage and the severity of the disease could not be made. Also, the travel status, reinfection could not be retrieved from the data which also led to failing of defining any relationship between the predominant lineage to be established.
On the basis of our study, we conclude that the various Omicron variant of different clades such as 21L, 22B, 22D and circulating BA.2, BA.2.38 and BA.2.75 were majorly circulating amongst the population of Uttarakhand and these had characteristic mutations in NTD such as T19I and V213G and in RBD such as S373P, S375F, T376A, and D405N. From the NGS, different mutations could be identified in fusion peptide, in spike protein and in furin cleavage site which led to identification of the reason why the Omicron variant was so transmissible than another variant. Therefore, to shed light on viral mutational insight which can affect its transmission and severity index, regular genomic surveillance done periodically is needed.
ACKNOWLEDGMENTS
The authors acknowledge the Indian Council of Medical Research-Department of Health Research (ICMR-DHR), New Delhi, India, for the financial help extended to set up a Viral Research and Diagnostic Laboratory (VRDL) at Government Doon Medical College, Dehradun, Uttarakhand, India. We would also like to acknowledge the help extended by Indian SARS-CoV-2 Genomics Consortium (INSACOG) for identification and confirmation of our sequencing data.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
AUTHORS’ CONTRIBUTION
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
FUNDING
None.
DATA AVAILABILITY
All datasets generated or analyzed during this study are included in the manuscript.
ETHICS STATEMENT
This study was approved by the Institutional Ethics Committee, Government Doon Medical College & Hospital, Dehradun, India, with reference number GDMC/IEC/2023/38.
- WHO. Statement on the second meeting of the International Health Regulations. Emergency Committee regarding the outbreak of novel coronavirus (2019-nCoV). 2005 https://www.who.int/news/item
- Saxena SK, Kumar S, Ansari S, et al. Characterization of the novel SARS-CoV-2 Omicron (B.1.1.529) variant of concern and its global perspective. J Med Virol. 2022;94(4):1738-1744.
Crossref - Jung C, Kmiec D, Koepke L, et al. Omicron: What Makes the Latest SARS-CoV-2 Variant of Concern So Concerning? J Virol. 2022;96(6):e0207721.
Crossref - Dai L, Gao GF. Viral targets for vaccines against COVID-19. Nat Rev Immunol. 2021;21(2):73-82.
Crossref - Centers of Disease Control and Prevention. Science brief: Omicron (B.1.1.529) variant. Accessed December 6, 2021.
- European Centre for Disease Prevention and Control. Implications of the emergence and spread of the SARS-CoV-2 B.1.1. 529 variant of concern (Omicron), for the EU/EEA.2021. Accessed December 4, 2021. https:// www.ecdc.europa.eu/en/publications-data/threat-assessment-brief emergence-sars-cov-2-variant-b.1.1.529
- Rana R, Kant R, Huirem RS, Bohra D, Ganguly NK. Omicron Variant: Current Insights and Future Directions. Microbiol Res. 2022;265:127204.
- Centers for Disease Control and Prevention (CDC), Omicron Variant: What You Need to Know. (n.d.). https://www.cdc.gov/coronavirus/2019-ncov/variants/o micron-variant.html
- Azami NAM, Perera D, Thayan R, et al. SARS-CoV-2 genomic surveillance in Malaysia: displacement of B.1.617.2 with AY lineages as the dominant Delta variants and the introduction of Omicron during the fourth epidemic wave. Int J Infect Dis. 2022;125:216-226.
Crossref - Raju MK, Thangaraj JWV, Selvavinayagam TS, et al. Clinical profile of patients infected with suspected SARS-CoV-2 Omicron variant of concern, Tamil Nadu, India, December 2021-January 2022. Indian J Med Res. 2022;155(1):165-170.
Crossref - Garg R, Gautam P, Suroliya V, et al. Evidence of early community transmission of Omicron (B1.1.529) in Delhi- A city with very high seropositivity and past-exposure. Travel Med Infect Dis. 2022;46:102276.
Crossref - Zaman K, Shete AM, Mishra SK, et al. Omicron BA.2 lineage predominance in severe acute respiratory syndrome coronavirus 2 positive cases during the third wave in North India. Front Med. 2022;9:955930.
Crossref - Sharma RP, Gautam S, Sharma P, et al. Genomic profile of SARS-CoV-2 Omicron variant and its correlation with disease severity in Rajasthan. Front Med. 2022;9:888408.
Crossref - Potdar VA, Yadav PD, Lole K, et al. Detection of the Omicron variant in international travellers and their family contacts in India. medRxiv. 2021.
Crossref - Ranjan R. Omicron Impact in India: Analysis of the Ongoing COVID-19 Third Wave Based on Global Data. medRxiv. 2022.
Crossref - INSACOG. INSACOG Bulletin. 2022:1-2. https://dbtindia. gov.in/insacog. 2022.
- WHO. Tracking SARS-CoV-2 Variants. 2022. Available online at: https://www.who. int/activities/tracking-SARSCoV-2-variants
- BA.2.75 – new Covid variant detected in India a mystery, but could ‘have immune-escape property. Available from: https://theprint.in/health/ba2-75-new-covid-variant-detected-in-india-A-mystery-but-could-have-immune-escape-property/1020578/.
- Martin DP, Lytras S, Lucaci AG, et al. Selection analysis identifies unusual clustered mutational changes in Omicron lineage BA.1 that likely impact Spike function. Preprint. bioRxiv. 2022.
Crossref - Chen KK, Tsung-Ning Huang D, Huang LM. SARS-CoV-2 variants – Evolution, spike protein, and vaccines. Biomed J. 2022;45(4):573-579.
Crossref - Sun C, Xie C, Bu GL, Zhong LY, Zeng MS. Molecular characteristics, immune evasion, and impact of SARS-CoV-2 variants. Signal Transduct Target Ther. 2022;7(1):202.
Crossref - Starr TN, Greaney AJ, Hilton SK, et al. Deep Mutational Scanning of SARS-CoV-2 Receptor Binding Domain Reveals Constraints on Folding and ACE2 Binding. Cell. 2020;182(5):1295-1310.e20.
Crossref - Li Q, Wu J, Nie J, et al. The Impact of Mutations in SARS-CoV-2 Spike on Viral Infectivity and Antigenicity. Cell. 2020;182(5):1284-1294.e9.
Crossref - Korber B, Fischer WM, Gnanakaran S, et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell. 2020;182(4):812-827.e19.
Crossref - Khandia R, Singhal S, Alqahtani T, et al. Emergence of SARS-CoV-2 Omicron (B.1.1.529) variant, salient features, high global health concerns and strategies to counter it amid ongoing COVID-19 pandemic. Environ Res. 2022;209:112816.
Crossref - Saito A, Tamura T, Zahradnik J, et al. Virological characteristics of the SARS-CoV-2 Omicron BA.2.75 variant. Cell Host Microbe. 2022;30(11):1540-1555.e15.
Crossref - Worldometer. India COVID coronavirus statistics. Available from: https://www.worldometers.info/coronavirus/country/india/.
- Hachmann NP, Miller J, Collier AY, et al. Neutralization Escape by SARS-CoV-2 Omicron Subvariants BA.2.12.1, BA.4, and BA.5. N Engl J Med. 2022;387(1):86-88.
Crossref - Cao Y, Yu Y, Song W, et al. Neutralizing antibody evasion and receptor binding features of SARS-CoV-2 Omicron BA.2.75. bioRxiv. 2022.
Crossref
© The Author(s) 2024. Open Access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License which permits unrestricted use, sharing, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.