


ISSN: 0973-7510
E-ISSN: 2581-690X
Phylogenetic analysis of bacteria and archaea on the seafloor surface of the Nankai Trough was conducted. DNA was extracted from a total of 14 samples (2 samples from 1 methane seep area and 12 samples from 12 general seafloor areas) and analyzed, targeting the 16S rRNA gene. As a result of the phylogenetic analysis of bacteria, 1 clone was found to have 96% homology with the 16S rRNA gene sequence of Methylomicrobium alcaliphilum, a halophilic methane-oxidizing bacterium, in a sample taken from the methane seep area. However, overall, no characteristic pattern was observed in the bacterial community structure between the methane seep area and the general seafloor. In contrast, in archaea, the genus Methanosarcina was predominantly detected in the samples from the methane seep area. Among them, many sequences that were closely related to anaerobic methane-oxidizing archaea, which perform anaerobic methane oxidation, were detected.
Methane seep, ANME, methane oxidation, MOB.
In the sea around Japan, it is known that there is a sand–mud alternating layer structure in the deep seafloor at a depth of 1,000–2,000 m, and methane hydrate is known to exist under the sand layer1,2. In the Nankai Trough, located off the southwest coast of Japan, the geological phenomenon is confirmed which related to the presence of methane hydrate under the seafloor, and observations using bottom-simulating reflectors showing the hydrate layer confirm this2,3. Methane hydrate is a clathrate hydrate; it is an ice-like solid substance formed only at low temperature and high pressure when numerous water molecules surround methane molecules. If we develop a technology that enables us to extract methane from methane hydrate, it would become a secure Japan’s original natural gas resources independent of import and expect development as a domestic energy resource.
In the future, methane gas may leak into seawater when recovering it from the methane hydrate layer in the deep seafloor because of the influence of ground deformation. Leakage of methane gas is a concern not only because of the loss of domestic energy source but also because of the effect that leakage could have on the ocean floor ecosystem. Therefore, the development of technology that prevents damage by detecting methane leakage at the bottom of the sea is considered to be an important issue in obtaining public acceptance.
Methane leakage can be defined as “a state in which methane exceeding a certain concentration is maintained for a certain period.” To investigate the effects of methane leakage on an ecological system, a group of Calyptogena on the southern slope of the Daini-Tenryu Knoll has been studied4,5. Calyptogena, known as deep-sea cold-seep clam, has been observed in cold-seep or hydrothermal vent and one of the representative organism in the cold-seep communitites6. It is presumed that methane leakage has some effect on the microbial community structures of an ecosystem because it causes a change in the surrounding environment.
Under aerobic condition, microbial oxidation of methane is well studied and aerobic methane-oxidizing bacteria (MOB), such as the genera Methylobacter, Methylococcus, and Methylosinus, are involved in the metabolism of methane7,8. Under the anaerobic condition, anaerobic oxidation of methane (AOM) is occurred. AOM has been reported that it is syntrophic reaction of sulfate-reducing bacteria and anaerobic methanotrophs called anaerobic methanotrophic (ANME) archaea9-12. These methane metabolism-related microbial communities can grow using methane as a electron donor, if the methane concentration in the surrounding environment rises due to methane leakage from the seafloor, and they can become a dominant microorganism group in that environment. Incidentally, regarding aerobic MOB, there are only few cases where they have been isolated and cultured from the deep seafloor13,14, but there are many cases where their existence has been confirmed at the genetic level15-18. The monitoring of the microorganisms that are characteristic of the methane seep area and the use of them as indicators can therefore help determine the levels of methane leakage.
PCR can be used to detect and analyze unknown microorganisms that have not been isolated previously, by performing phylogenic analysis of target genes18,19.
Therefore, in this study microbial community in the surface layer of the ocean floor was analyzed based on 16S rRNA gene sequence. In this paper, samples of submarine mud surface were collected in the methane seep and general seafloor areas (control) from the surface layer sediments of the Nankai Trough. We then investigated the composition and characteristics of the microbial ecosystems to determine whether differences exist between methane seepage and control areas.
Sampling sites and methods
In October-November 2004, sediments on the sea floor from 13 different locations were collected during the voyage of Bosei-maru, owned by Tokai University. The sampling sites were targeted at the Nankai Trough area located to the east of the Kumano-nada (Table 1). Here, the seafloor surface layer sample (S1) from the methane seep area was set as the target area where methane was present, and the other 12 locations were investigated as nonmethane-existence areas. S1 is the point where the colony of Calyptogena was observed19, and the remaining were general seafloor sediments.
In the marine sediment of S1, surface mud (approximately 20 cm) of the seafloor was collected, together with the group of Calyptogena, using a grab sampler (Kinoshita-type sampler, equipped with a seafloor camera). The surface of S1 sample was gray, but the central part of the sediment was black. The black part was designated as “reductive sample, S1k,” and the remaining part was designated as “oxidative sample, S1s.” These 2 samples were investigated. The remaining 12 samples (MC01, MC02, MC03, MC04, MC05, MC06, PC01, PC03, PC04, PC09, PC10, PC11) were general seafloor samples taken as controls for comparison with samples from the methane seep area. For MC01, MC02, MC03, MC04, MC05, and MC06, the surface layer part (approximately 30 cm) of the seabed was sampled in an undisturbed manner using a multiple corer. Columnar surface sediments (from the seafloor surface to approximately 5 m below) were collected for PC01, PC03, PC04, PC09, PC10, and PC11 using a piston corer (pipe length: 5 m, with an Ewing-type pilot corer).
Among the marine sediments collected using the multiple corer or piston corer, approximately 10 ml of pore water was extracted from about 200 g of sediments, using hydraulic pressure, for chemical analysis. A portion (approximately 2 ml) of the extracted pore water was collected in a 2-ml vial bottle containing mercuric chloride and amidosulfuric acid, and the bottles were tightly sealed for the analysis of gas components. For dissolved methane analysis, surface mud sample of S1 site and sea water samples just above the seafloor (ca. 10 cm) of the other site were used.
Dissolved methane concentration
The dissolved methane concentration was measured by the purge method because the concentration was predicted to be too low in the pore water sample20. The measurement of the methane concentration in the sediment sample was performed according to the method of Tsunogai et al.21, using a continuous-flow isotope ratio mass spectrometer by the headspace method.
Phylogenetic analysis
DNA was extracted from 1.5–2.0 g of the ocean sediment using an Ultra Clean Soil DNA Kit (Mo Bio Laboratories) and ISOIL (Nippon Gene) and mixed in equal amounts. This DNA solution was used for PCR to amplify the 16S rRNA gene of bacteria and archaea. For the PCR, thermal cycler GeneAmp 9600 (Perkin Elmer) and Ampli Taq Gold (Applied Biosystems) were used. The PCR reaction was performed under the same conditions for both archaea and bacteria at 94°C for 10 minutes followed by 35 cycles at 94°C for 1 minute, 58°C for 1 minute, and 72°C for 2 minutes. After 35 cycles, the samples were held at 72°C for 10 minutes. For primer pairs, 8F and 1492R were used for the amplification of the bacterial 16S rRNA gene22, and ARC344F and 1492R were used for the amplification of the archaeal 16S rRNA gene23.
After confirming the fragment size by agarose gel electrophoresis, the obtained PCR product was excised from the gel, extracted, and purified using a Gel Extraction Kit (QIAGEN, Hilden, Germany). Thereafter, it was cloned into the TA Cloning vector pCR 2.1 and transfected into an One Shot TOP10 competent cell (Invitrogen, Carlsbad, San Diego, CA). The sequencing was performed with 310 Genetic Analyzer (Applied Biosystems, Foster City, CA). The obtained sequence was checked for homology with nucleotide sequences on the NCBI databases, and after alignment with the ClustalW program24, a phylogenetic tree was constructed by the neighbor-joining method.
To compare the microbial community structure in each sample, principal component analysis (PCA) was performed. For the analysis, we used the detection rate of the phylum and class level classification of each sample; STAMP software25 was used for calculations based on default setting of Euclidean distance.
The 16S rRNA gene sequence data obtained in this study and used for phylogenetic analysis were deposited in the GenBank/EMBL/DDBJ databases under accession numbers LC382285 to LC382364.
Dissolved methane concentration
The methane concentration in the sediment sample of the general seafloor was approximately 0.25µmol/L, which is the background level in the seafloor of this survey area. Because the methane seep area was inhabited by a group of Calyptogena, the corer could not penetrate and the sediment sample was collected using a grab-type sampler. Therefore, the dissolved methane concentration was not measured.
Phylogenic analysis of bacterial 16S rRNA gene sequence
In total, 1,038 bacterial clones were sequenced for the 16S rRNA gene. Most of the clones obtained from each sample were classified into the phyla Proteobacteria or Bacteroidetes, which are Cytophaga and Flavobacterium (Fig. 1). Although the ratio of the dominant clone was different for each site, g-Proteobacteria, d-Proteobacteria, and Bacteroidetes were detected at all sites. The proportion of g-Proteobacteria, which belongs to type I of aerobic methane-oxidizing bacteria, was overwhelmingly 47.5% in PC09, followed by PC10 (35.7%) and PC11 (32.4%). In addition, d-Proteobacteria, to which many sulfate-reducing bacteria belong, was detected at all sites. The largest percentage was 37.2% for MC02, followed by 25.0% for MC05. e-Proteobacteria, to which many sulfur-oxidizing bacteria belong, were detected in PC03, and the proportion accounted for 35.9%. Although a-Proteobacteria were not detected at some sites, a relatively large proportion was detected in PC04 and PC11 (35.6% and 35.1%, respectively).
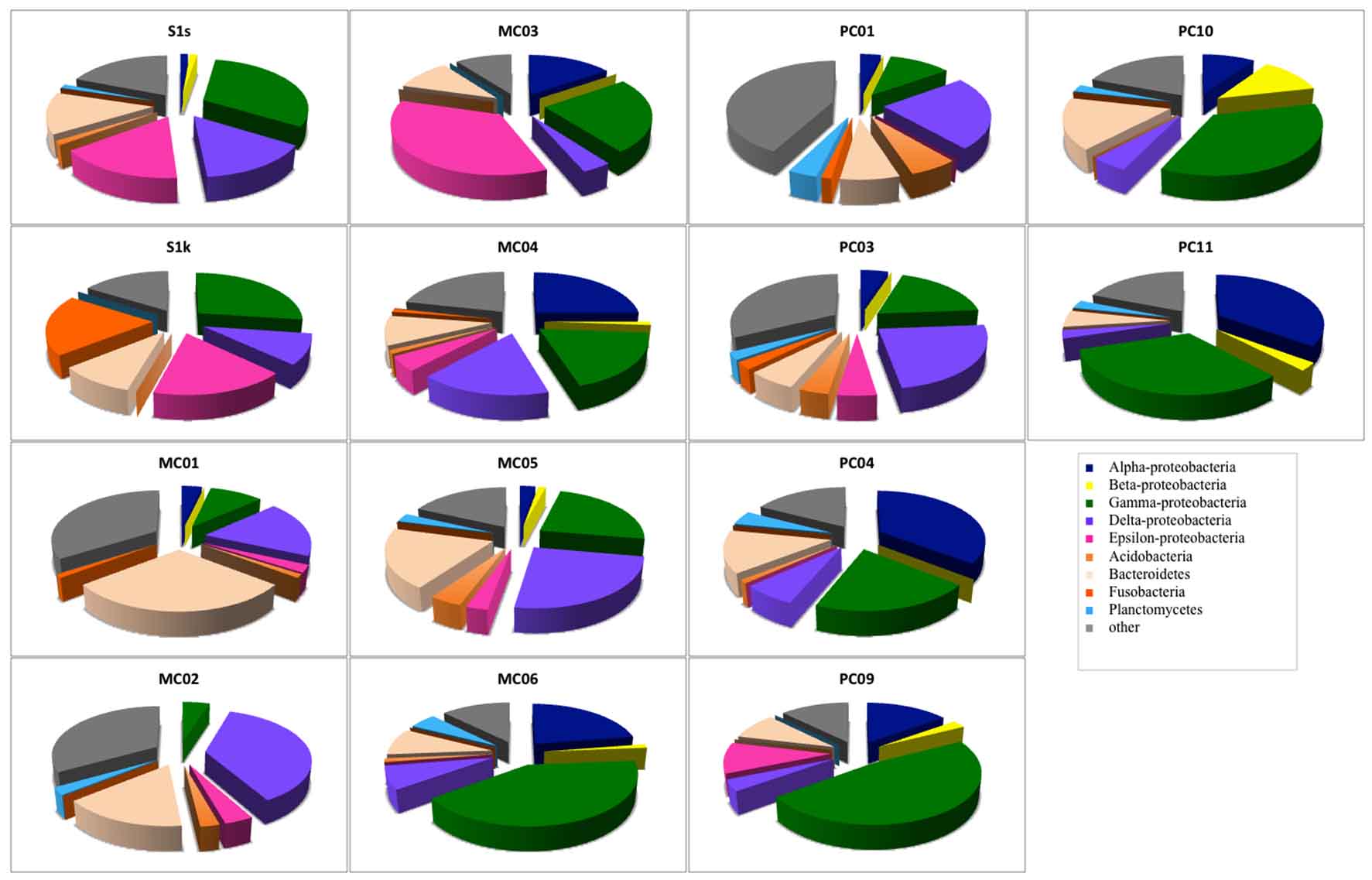
Fig. 1. Bacterial community compositions of marine sediments based on 16S rRNA gene sequence analysis.
Analysis of each site showed that there were many g-Proteobacteria (approximately 30%) in S1s and S1k that were from the methane seep area.
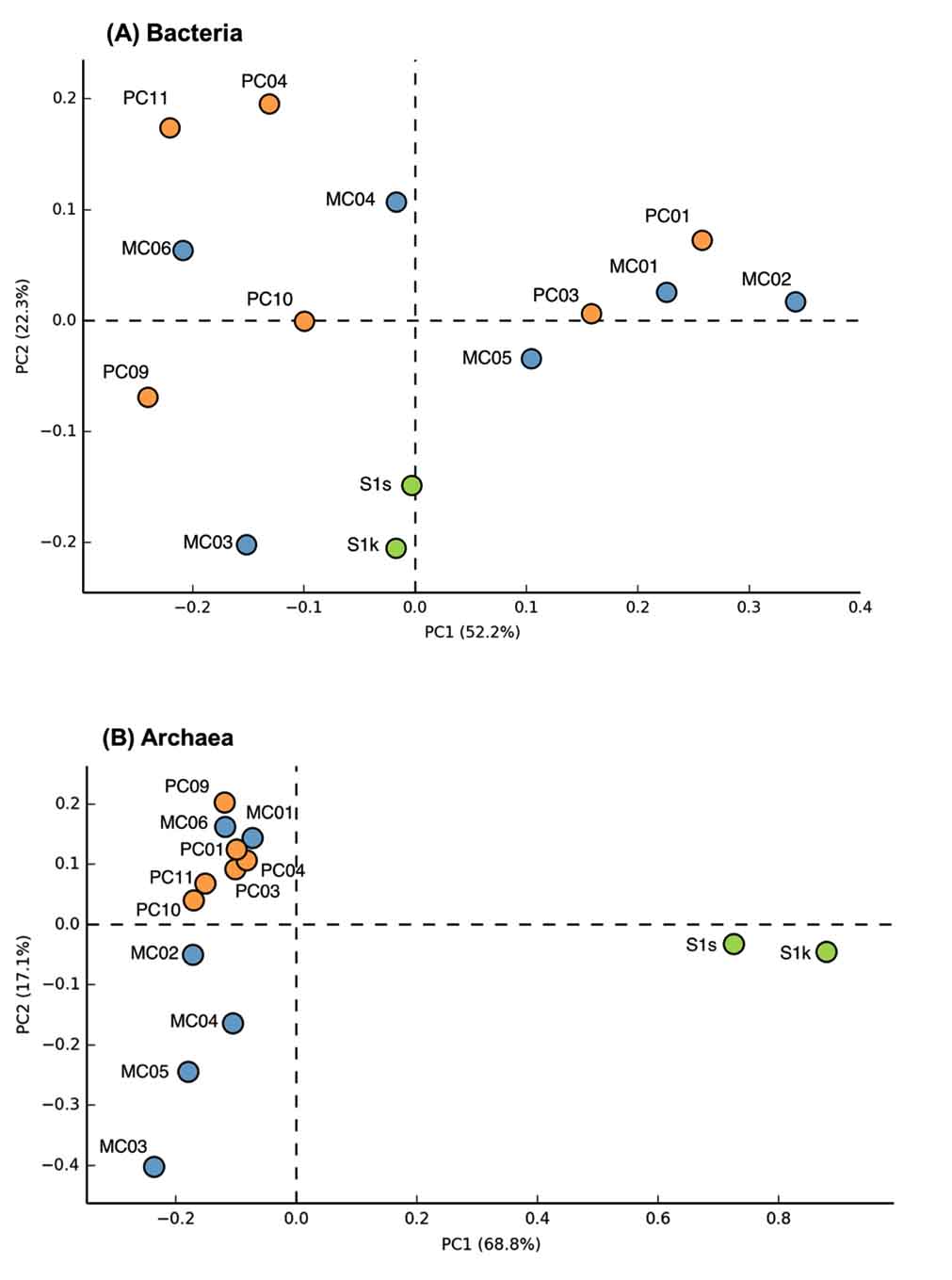
Fig. 2. Principal component analysis plots of bacterial (A) and archaeal (B) communities of marine sediments.
The groups that most dominated in the mud sample of the general seafloor were as follows; MC01: Bacteroidetes (29.3%), MC02: d Proteobacteria (37.0%), MC03: e-Proteobacteria (35.9%), MC04: a-Proteobacteria (25%), MC05: d-Proteobacteria (25.0%), MC06: g-Proteobacteria (41.3%), PC01: d-Proteobacteria (23.8%), PC03: d-Proteobacteria (23.8%), PC04: a-Proteobacteria (35.6%), PC09: g-Proteobacteria (47.5%), PC10: g-Proteobacteria (35.7%), PC11: a-Proteobacteria (35.1%). Thus, the microflora exhibited different compositions for each site, and when compared with the 16S rRNA gene sequence of bacteria as an index, a significant pattern, capable of discriminating between the methane seep and general seafloor areas, was not observed. The same result was obtained by PCA based on the detection ratio of each bacterial group. However, it was found that the 2 samples in the methane seep area were similar to each other (Fig. 2A).
Nearly half of the sequenced clones demonstrated low homology (<95%) with strains and genes from the database and were derived from microorganisms that were completely unknown at the genus level. In almost all the samples, no distinction was made between whether the sample was from the methane seep or general seafloor areas. Clones with high homology with clones collected from the methane seep area26,27 in the sea around Japan and other oceanic gas hydrate28,29 and hydrocarbon seep areas30 were obtained (³97%). In addition, clones related to methane oxidation (closely related to the methanotrophic community) were obtained from 8 samples of S1s, S1k, MC01, MC02, MC03, MC04, PC01, and PC03. However, in our study, clones corresponding to the known methanotrophic bacteria were not obtained, and only 1 clone with 96% homology with Methylomicrobium alcaliphilum31, a halophilic methane-oxidizing bacterium, was found in a sample from the methane seep area.
Phylogenic analysis of archaea 16S rRNA gene sequence
In the archaea 16S rRNA gene, 1, 105 clones were sequenced. As a result of the analysis of archaea, a remarkable difference was observed between the methane seep and general seafloor areas (Figs. 2B and 3). From the S1s sample, 76 clones were sequenced, 84% of which were closely related to the genus Methanosarcina. From the S1k sample, the 80 clones obtained were classified into 2 phylogenies, Methanosarcina (79 clones) and DSAG (1 clone). Thus, the genus Methanosarcina overwhelmingly dominated in the methane seep area, whereas there were no sites dominated by the genus Methanosarcina in the other 12 general seafloor samples.

Fig. 3. Archaeal community compositions of marine sediments based on 16S rRNA gene sequence analysis.
In the 12 general seafloor samples, it was found that each site has a unique composition at the genus level. When analyzing the dominant group at each site, it was found that MC01 is dominated by the marine benthic group-E (MBGE) and marine benthic group-D (MBGD) (22% and 20%, respectively). The clones obtained in MC02, MC03, MC04, and MC05 were dominated by the 10-PML 8% pond group (24%, 60%, 35% and 42%, respectively), which is composed of clones obtained from saline soil and solar saltern with homology as low as 90%. MC06 was dominated by MBGD (22%), PC01 was dominated by MBGE (23%), and PC03 was dominated by MBGD (35%). In PC04, MBGE accounted for 19%, and the pBRKC 84 group32 (formed by clones with homology as low as 90% with clones obtained from hypersaline microbial mats) accounted for 18%. PC09 was dominated by MBGD (33%). In PC10 and PC11, MBGD dominated (25% and 38%, respectively), followed by 10-PML 8% pond group (19% and 17%, respectively).
Similar to the bacterial results, many archaea clones obtained in this study were not matched with any strain or gene clone from the database (<95%) and were derived from microorganisms that are completely unknown at the genus level. Similar to the bacterial results, in all samples from the general seafloor area, clones with high homology with clones collected from the methane seep area in the sea around Japan and other oceanic gas hydrate and hydrocarbon seep areas were obtained (³97%). Clones showing high homology (³97%) with clones involved in methane oxidation (closely related to the methanotrophic community) were obtained from general seafloor samples other than PC01 and PC10.
To further analyze the genus Methanosarcina obtained from S1, the phylogenetic relationship was analyzed by the sequence of the 16S rRNA gene sequence classified in the genus Methanosarcina. Consequently, it was found that 52 of 64 genus Methanosarcina clones obtained from S1s are closely related to ANME-2, and some clones were closely related to ANME-3 belonging to the genus Methanococcoides. In contrast, among 79 clones of the genus Methanosarcina obtained from S1k, 51 were closely related to ANME-2 and 27 were closely related to ANME-3 (Fig. 4).
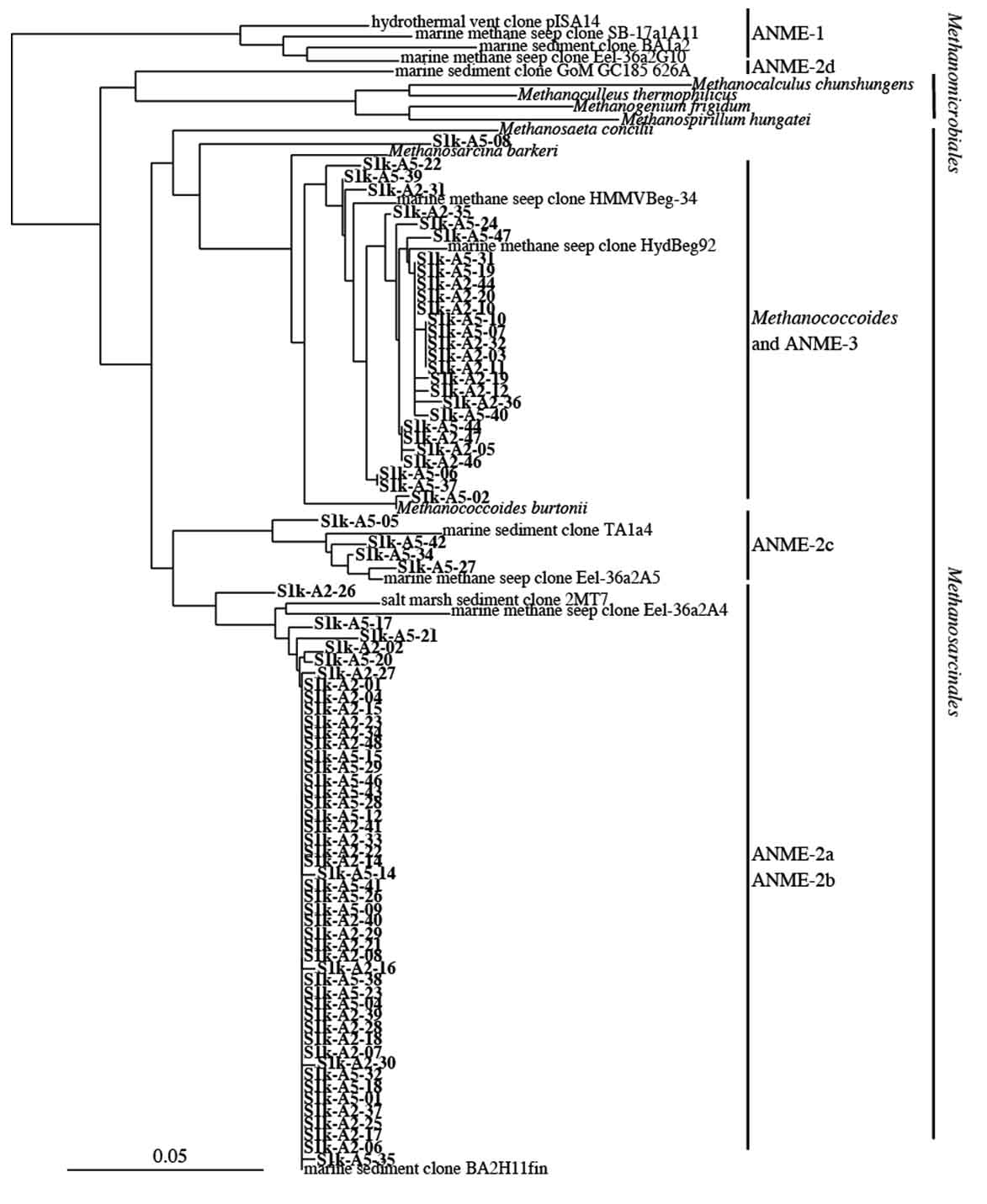
Fig. 4. Phylogenetic analysis of archaeal 16S rRNA gene sequences obtained from S1 site sample. The scale bar represents the number of nucleotide changes per sequences position.
Although methane concentration in the ocean is <0.5 nmol/l33, in some special ocean areas, it exceeds 5 nmol/l34. Areas in the sea have been identified where methane is generated by microorganisms from sludge sediments (e.g., in Tokyo Bay, where organic matter has become sludge) and where methane is released by thermal generation from cold-water seep areas and hydrothermal polymetallic ores of the open sea. For example, it has been reported that methane concentration is 340 לmol/kg in pore water in sediments of cold-water seep areas (depth, 1,100 m), which contain methane hydrates, of the Ryuyo Canyon in the eastern Nankai Trough area35. In the present study, the methane concentration in the pore water could not be measured in the cold-water seep area (S1 site, Table 1) of the Daini Tenryu Knoll, selected in the survey cruise of the eastern Nankai Trough area in 2004. However, the fact that unusual organisms inhabit the methane seep area, such as populations of Calyptogena, demonstrates that an ecosystem that is clearly different from other low-methane-concentration seafloors exists.
Table (1):
Locations of sample collection sites .
Sample name |
Latitude |
Longitude |
Water depth (m) |
note |
|---|---|---|---|---|
S1 |
34°04.594’N |
137°47.265’E |
611 |
Methane seep |
MC01 |
33°55.770’N |
137°18.902’E |
984 |
|
MC02 |
33°49.005’N |
136°30.353’E |
2,053 |
|
MC03 |
33°59.983’N |
137°10.225’E |
1,852 |
|
MC04 |
34°08.740’N |
137°29.415’E |
1,359 |
|
MC05 |
34°12.230’N |
137°27.615’E |
1,258 |
|
MC06 |
34°16.557’N |
137°57.921’E |
734 |
|
PC01 |
33°57.649’N |
137°15.829’E |
1,440 |
|
PC03 |
33°49.164’N |
136°30.056’E |
2,053 |
|
PC04 |
33°50.734’N |
136°29.658’E |
1,886 |
|
PC09 |
34°13.346’N |
137°40.265’E |
1,166 |
|
PC10 |
34°16.587’N |
137°44.841’E |
922 |
|
PC11 |
34°13.145’N |
137°42.702’E |
1,076 |
A group of microorganisms called methanotrophs is known to be involved in methane conversion and are related to methane oxidation in an aerobic environment6,7. Based on this, we can predict that aerobic MOB will be detected in the S1 sample that is collected from a site where methane seeps from the sea floor; however, known aerobic methane-oxidizing bacteria were not detected. This is because 16S rRNA is a gene common to all microorganisms, it was considered that it is not appropriate to target bacterial 16S rRNA as a detection method focusing on aerobic MOB only.
Interestingly, compared with the sequence analysis results of other studies, we found clones showing high homology (e”97%) with sequences presumed to be involved in methane oxidation (members of the methanotrophic community) in the samples, not only from the methane seep area (S1) but also from the general seafloor area in the Nankai Trough10, 36. The microorganism possessing these sequences yet to be isolated and cultured, thus it is unknown whether these microorganisms can utilize methane in nature. However, it has been suggested that microorganisms capable of utilizing methane are widely distributed in the surface layer of the ocean floor. Thus, it can be considered that there exists a microbial community universally involved in methane oxidation in the mud of the seafloor, and if methane is supplied from the bottom of the sea, a potential methane utilization community may be constructed.
Many studies have found that ecological systems exist in deep sea mud volcanoes and methane seep areas; these ecological systems are supported by chemolithotrophs that depends on methane and sulfuric acid, and anaerobic methane oxidation occurs in places where methane and sulfuric acid coexist. Therefore, the process of anaerobic methane oxidation is thought to be associated with ANME archaea and sulfate-reducing bacteria (SRB)9,10,37,38, and a report has stated that ANME-2 has a close relationship with SRB genera, Desulfosarcina and Desulfococcus9,39,40. In addition, in the present study, because clones closely related to ANME-2 were detected in the samples from methane seep areas, we focused on clones of d-Proteobacteria, to which many SRB belong. However, clones corresponding to or closely related to the known genera Desulfosarcina and Desulfococcus were not detected.
Microbial community analysis, based on archaeal 16S rRNA gene sequence, showed a remarkably different microbial community structure between samples from the methane seep and general seafloor areas (Fig. 2B). In the S1 methane seep area, the genus Methanosarcina dominated overwhelmingly, and the diversity of the microbial flora was extremely low. This result was similar to other reported results of methane-rich sediment samples12,41,42. Furthermore, the S1 methane seep area community contained many clones closely related (97% to 99%) to the ANME group that perform anaerobic methane oxidation. ANME has also been detected in several other methane seep areas investigated in previous studies43.
In culture experiments conducted by Girguis et al., anaerobic methane oxidation activity and sequences of the ANME-2 group were detected after culturing with methane44. Therefore, their results support the speculation obtained from the results of our analysis. In general submarine environments that consist of various microbial community structures, dominance of the Methanosarcina seen in sample S1 is believed to be the result of a change in the microbial flora along with the environmental change of the methane seep area. From this, it was considered that a sequence of the ANME group could be one of the characteristic microbial indices on the seafloor where methane exists.
In our analysis, sequences closely related to ANME-2 and ANME-3 were detected in large amounts in the S1 site, which is a methane seep area. It has been reported that ANME-1, another group involved in anaerobic methane oxidation, are detected and are present specifically in methane seep areas8. Although it was previously confirmed that the detection of ANME-1 with the primer pair used in our study is possible, ANME-1 was not detected at all in our analysis. Moreover, it has been proven that ANME-1 exists in the deep ocean floor45. Because the water depth of the S1 site analyzed in our study was approximately 600 m, there is a possibility that it is not the habitat of ANME-1.
In the marine mud where methane hydrate is present, it is known that only moisture from saltwater is taken up by methane hydrate, and the salt concentration increases in the surrounding area. In the phylogenetic analysis of archaea, although the homology was low, many clones were obtained that form the same branches as the sequences obtained from areas with extremely high salt concentrations. In addition, clones closely related to halophilic methane-oxidizing bacteria were obtained in the phylogenetic analysis of bacteria. It is interesting to observe the relationship between these results and methane hydrate.
In conclusion, generally, it is highly likely that a unique microbial flora is formed on the seafloor where methane is released, and this analysis suggests that it is not an exception in the Nankai Trough. By targeting genes of microorganisms that rapidly respond to methane leakage, it is possible to detect methane leakage on the seafloor.
ACKNOWLEDGMENTS
We would like to thank Mr. Shunji Sukizaki for collecting the samples.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
AUTHORS’ CONTRIBUTION
T.H. and S.S. designed research; T.H. and S.S. performed sample collection; N.O., T.H. and M.H. analyzed data; N.O., M.H. and T.H. wrote the manuscript.
FUNDING
This work was a part of MH21 research consortium activities funded by the Ministry of Economy, Trade and Industry (METI), Japan.
ETHICS STATEMENT
This article does not contain any studies involving human participants and animals performed by any of the authors.
AVAILABILITY OF DATA
Sequence data obtained in this study were available in GenBank/EMBL/DDBJ databases at accession numbers LC382285 to LC382364.
- Research Consortium for Methane Hydrate Resources in Japan. Phase 1 Comprehensive Report of Research Results. 2008.
- Colwell F, Matsumoto R, Reed D. A review of the gas hydrates, geology, and biology of the Nankai Trough. Chemical Geology., 2004; 205(3): 391-404.
Crossref - Uchida T, Lu H, Tomaru H. Subsurface Occurrence of Natural Gas Hydrate in the Nankai Trough Area: Implication for Gas Hydrate Concentration. Resource Geology, 2004; 54(1): 35-44.
Crossref - Kuramoto S. Possibility of intermittent emission of methane gas. Chikyu Monthly. 2001; 32: 130-5.
- Miyazaki J. Microbial Ecology and Metabolisms in Methane-seep Sites Related to Mud Volcanoes. Journal of Geography (Chigaku Zasshi). 2009; 118(3): 564-77.
Crossref - Mizota, C., Maki, Y.. Sulfur isotopic variations in soft tissues of Calyptogena soyoae from Sagami Bay, central Japan. Geochemical Journal, 1998; 32(6): 421-425.
Crossref - Hanson RS, Hanson TE. Methanotrophic bacteria. Microbiological Reviews, 1996; 60(2): 439-71.
- George Ge. Bergey’s Manual of Systematic Bacteriology. New York: Springer US; 2005.
- Orphan VJ, Hinrichs K-U, Ussler W, Paull CK, Taylor LT, Sylva SP, et al. Comparative Analysis of Methane-Oxidizing Archaea and Sulfate-Reducing Bacteria in Anoxic Marine Sediments. Applied and Environmental Microbiology, 2001; 67(4): 1922-34.
Crossref - Teske A, Hinrichs K-U, Edgcomb V, de Vera Gomez A, Kysela D, Sylva SP, et al. Microbial Diversity of Hydrothermal Sediments in the Guaymas Basin: Evidence for Anaerobic Methanotrophic Communities. Applied and Environmental Microbiology, 2002; 68(4): 1994-2007.
Crossref - Boetius A, Ravenschlag K, Schubert CJ, Rickert D, Widdel F, Gieseke A, et al. A marine microbial consortium apparently mediating anaerobic oxidation of methane. Nature, 2000; 407: 623.
Crossref - Hinrichs K-U, Hayes JM, Sylva SP, Brewer PG, DeLong EF. Methane-consuming archaebacteria in marine sediments. Nature, 1999; 398: 802.
Crossref - Nakamura T, Hoaki T, Hanada S, Maruyama A, Kamagata Y, Fuse H. Soluble and particulate methane monooxygenase gene clusters in the marine methanotroph Methylomicrobium sp. strain NI. FEMS Microbiology Letters, 2007; 277(2): 157-64.
Crossref - Fuse H, Ohta M, Takimura O, Murakami K, Inoue H, Yamaoka Y, et al. Oxidation of Trichloroethylene and Dimethyl Sulfide by a Marine Methylomicrobium Strain Containing Soluble Methane Monooxygenase. Bioscience, Biotechnology, and Biochemistry, 1998; 62(10): 1925-31.
Crossref - Duperron S, Nadalig T, Caprais JC, Sibuet M, Fiala-Medioni A, Amann R, et al. Dual Symbiosis in a Bathymodiolus sp. Mussel from a Methane Seep on the Gabon Continental Margin (Southeast Atlantic): 16S rRNA Phylogeny and Distribution of the Symbionts in Gills. Applied and Environmental Microbiology, 2005; 71(4): 1694.
Crossref - Duperron S, Sibuet M, MacGregor BJ, Kuypers MMM, Fisher CR, Dubilier N. Diversity, relative abundance and metabolic potential of bacterial endosymbionts in three Bathymodiolus mussel species from cold seeps in the Gulf of Mexico. Environmental Microbiology, 2007; 9(6): 1423-38.
Crossref - Tchawa Yimga M, Dunfield PF, Ricke P, Heyer J, Liesack W. Wide Distribution of a Novel <em>pmoA</em>-Like Gene Copy among Type II Methanotrophs, and Its Expression in <em>Methylocystis</em> Strain SC2. Applied and Environmental Microbiology, 2003; 69(9): 5593-602.
Crossref - Reed AJ, Dorn R, Van Dover CL, Lutz RA, Vetriani C. Phylogenetic diversity of methanogenic, sulfate-reducing and methanotrophic prokaryotes from deep-sea hydrothermal vents and cold seeps. Deep Sea Research Part II: Topical Studies in Oceanography, 2009; 56(19): 1665-74.
Crossref - Hoaki T, Okita N, Kunii N, Sukisaki S, Yoshida K, Fuse H. Search for microbial genetic marker in surface sediment of seafloor: development of in-situ detection instrumentation technology for the leakage methane. Kaiyo monthly. 2006; 38(12): 862-87. [In Japanese]
- Tsunogai U, Yoshida N, Ishibashi J, Gamo T. Carbon isotopic distribution of methane in deep-sea hydrothermal plume, Myojin Knoll Caldera, Izu-Bonin arc: implications for microbial methane oxidation in the oceans and applications to heat flux estimation. Geochimica et Cosmochimica Acta., 2000; 64(14): 2439-52.
Crossref - Tsunogai U, Ishibashi J, Wakita H, Gamo T. Methane-rich plumes in the Suruga Trough (Japan) and their carbon isotopic characterization. Earth and Planetary Science Letters, 1998; 160(1): 97-105.
Crossref - Hendrickson ER, Payne JA, Young RM, Starr MG, Perry MP, Fahnestock S, et al. Molecular Analysis of <em>Dehalococcoides</em> 16S Ribosomal DNA from Chloroethene-Contaminated Sites throughout North America and Europe. Applied and Environmental Microbiology, 2002; 68(2): 485-95.
Crossref - Casamayor EO, Schafer H, Baneras L, Pedros-Alio C, Muyzer G. Identification of and Spatio-Temporal Differences between Microbial Assemblages from Two Neighboring Sulfurous Lakes: Comparison by Microscopy and Denaturing Gradient Gel Electrophoresis. Applied and Environmental Microbiology, 2000; 66(2): 499-508.
Crossref - Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Research, 1994; 22(22): 4673-80.
Crossref - Parks DH, Tyson GW, Hugenholtz P, Beiko RG. STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics, 2014; 30(21): 3123-4.
Crossref - Fang J, Shizuka A, Kato C, Schouten S. Microbial diversity of cold-seep sediments in Sagami Bay, Japan, as determined by 16S rRNA gene and lipid analyses. FEMS Microbiology Ecology, 2006; 57(3): 429-41.
Crossref - Arakawa S, Sato T, Sato R, Zhang J, Gamo T, Tsunogai U, et al. Molecular phylogenetic and chemical analyses of the microbial mats in deep-sea cold seep sediments at the northeastern Japan Sea. Extremophiles : life under extreme conditions, 2006; 10(4): 311-9.
Crossref - Inagaki F, Nunoura T, Nakagawa S, Teske A, Lever M, Lauer A, et al. Biogeographical distribution and diversity of microbes in methane hydrate-bearing deep marine sediments on the Pacific Ocean Margin. Proceedings of the National Academy of Sciences of the United States of America, 2006; 103(8): 2815-20.
Crossref - Mills HJ, Martinez RJ, Story S, Sobecky PA. Characterization of Microbial Community Structure in Gulf of Mexico Gas Hydrates: Comparative Analysis of DNA- and RNA-Derived Clone Libraries. Applied and Environmental Microbiology, 2005; 71(6): 3235-47.
Crossref - LaMontagne MG, Leifer I, Bergmann S, Van De Werfhorst LC, Holden PA. Bacterial diversity in marine hydrocarbon seep sediments. Environmental Microbiology, 2004; 6(8): 799-808.
Crossref - Khmelenina VN, Kalyuzhnaya MG, Starostina NG, Suzina NE, Trotsenko YA. Isolation and Characterization of Halotolerant Alkaliphilic Methanotrophic Bacteria from Tuva Soda Lakes. Current Microbiology, 1997; 35(5): 257-61.
Crossref - Bowman JP, McCuaig RD. Biodiversity, Community Structural Shifts, and Biogeography of Prokaryotes within Antarctic Continental Shelf Sediment. Applied and Environmental Microbiology, 2003; 69(5): 2463-83.
Crossref - T. Gamo JI, K. Shitashima, M. Kinoshita, M. Watanabe, E. Nakayama, Y. Sohrin, E.-S. Kim, T. Masuzawa and K. Fujioka Anomalies of bottom CH4 and trace metal concentrations associated with high heat flow at the Calyptogena community off Hatsu-shima Island, Sagami Bay, Japan: A preliminary report of Tansei Maru KT-88-1 cruise Leg-1. Geochemical Journal, 1988; 22(5): 215-30.
Crossref - Boulegue J, Iiyama JT, Charlou J-L, Jedwab J. Nankai Trough, Japan Trench and Kuril Trench: geochemistry of Fluids sampled by submersible “Nautile”. Earth and Planetary Science Letters, 1987; 83(1): 363-75.
Crossref - Tsunogai U, Wakita H, Ashi J, Gamo T. Fresh Water seepage on the seafloor: Ryuyo Canyon, Nankai Trough. JAMSTEC J. Deep Sea Res., 1997; 13: 503-8.
- Losekann T, Knittel K, Nadalig T, Fuchs B, Niemann H, Boetius A, et al. Diversity and Abundance of Aerobic and Anaerobic Methane Oxidizers at the Haakon Mosby Mud Volcano, Barents Sea. Applied and Environmental Microbiology, 2007; 73(10): 3348.
Crossref - Lars Bille H, Kai F, Henrik F, Niels I. Anaerobic methane oxidation in sulfate depleted sediments: effects of sulfate and molybdate additions. Aquatic Microbial Ecology, 1998; 14(2): 195-204.
Crossref - Hoehler TM, Alperin MJ, Albert DB, Martens CS. Field and laboratory studies of methane oxidation in an anoxic marine sediment: Evidence for a methanogen-sulfate reducer consortium. Global Biogeochemical Cycles, 1994; 8(4): 451-63.
Crossref - Knittel K, Losekann T, Boetius A, Kort R, Amann R. Diversity and distribution of methanotrophic archaea at cold seeps. Applied and Environmental Microbiology. 2005; 71(1): 467-79.
Crossref - Orphan VJ, House CH, Hinrichs K-U, McKeegan KD, DeLong EF. Multiple archaeal groups mediate methane oxidation in anoxic cold seep sediments. Proceedings of the National Academy of Sciences, 2002; 99(11): 7663-8.
Crossref - Thomsen TR, Finster K, Ramsing NB. Biogeochemical and molecular signatures of anaerobic methane oxidation in a marine sediment. Applied and environmental microbiology. 2001; 67(4): 1646-56.
Crossref - Bidle KA, Kastner M, Bartlett DH. A phylogenetic analysis of microbial communities associated with methane hydrate containing marine fluids and sediments in the Cascadia margin (ODP site 892B). FEMS Microbiology Letters, 1999; 177(1): 101-8.
Crossref - Roalkvam I, Jorgensen SL, Chen Y, Stokke R, Dahle H, Hocking WP, et al. New insight into stratification of anaerobic methanotrophs in cold seep sediments. FEMS Microbiology Ecology, 2011; 78(2): 233-43.
Crossref - Girguis, P.R., Orphan, V.J., Hallam, S.J., DeLong, E.F. Growth and Methane Oxidation Rates of Anaerobic Methanotrophic Archaea in a Continuous-Flow Bioreactor. Applied and Environmental Microbiology, 2003; 69(9): 5472-5482.
Crossref - Schubert CJ, Coolen MJL, Neretin LN, Schippers A, Abbas B, Durisch-Kaiser E, et al. Aerobic and anaerobic methanotrophs in the Black Sea water column. Environmental Microbiology, 2006; 8(10): 1844-56.
Crossref
© The Author(s) 2019. Open Access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License which permits unrestricted use, sharing, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.