


Hepatitis C is widely spread and induces life threatening situations. Researchers from various fields have developed vaccine, but they are not that effective because of the variation in genotype of Hepatitis virus and also not much affordable. In-silico approach is of greater importance in designing and testing the model vaccine. In this study, investigation has been done for the available Bioinformatics tools and methodologies used in HCV research. Different types of tools and databases commonly used by researchers were reviewed to get an overall picture of bioinformatics techniques, computational biology tools and databases used in Hepatitis C Virus research. Exclusive study has also done to figure out different statistical methods used by different research groups. This paper will provide an up-to-date picture of computation approaches used for exploring Hepatitis C treatment.
Liver disease, In-silico, tools, techniques, database, vaccine.
The causative agent of Hepatitis C, a life threatening disease is Hepatitis C virus. Characteristically it affects the liver and the individual can develop acute and chronic infections. The infection starts with slight infirmity for few weeks to major lifetime illness. HCV infection occurs via the blood transfusion of the infected patient to the normal individual, using non sterilized medical equipment, using same syringe or needle for more than one individual. Globally 130–150 million people develop chronic hepatitis C infection1. Out of those, considerable numbers of chronically infected individuals develop liver cirrhosis and cancer. Numbers of individual who lose their lives from this infection are close to 500,000 per year2. Among the HCV infected population antiviral medicines can neutralise the effect of 90% population which leads to minimized chances of death from liver cirrhosis and cancer but access to diagnosis and treatment protocols are very poor. Research to avail first vaccine globally is still in process3. Hepatitis C virus came into picture in 1989 by expression cloning of immunoreactive cDNA isolated from infectious non-A and non-B Hepatitis agent. HCV comes under the flaviviridae family and genus Hepacivirus. Its genetic material comprises of positive single-stranded RNA. The genome size of HCV is 9.6 kb RNA having highly structured 5′ and 3′ ends (Fig. 1). The 5′ end is 341 nucleotides long conserved non coding region which contains four major domains when folded into complex structure. Flavivirus codes for a long open reading frame having 5′ cap and conserved RNA structures at both the 5′ and 3′ untranslated regions essential for replication and translation of viral genome4. The structural HCV proteins are currently designated as core, E1, E2 and p7. Additionally, the non-structural proteins are NS2, NS3, NS4A, NS4B, NS5A and NS5B. The genomic RNA is translated into a single polyprotein precursor consisting of three structural Capsid (C), perinuclear membrane (prM), and Envelop (E) protein and seven non-structural NS1, NS2a, NS2b, NS3, NS4a, NS4b and NS5 proteins arranged in the order C-prM-E-NS1-NS2a-NS2b-NS3-NS4a-NS4b-NS5. Mature and infectious virion contains only the structural proteins and the non-structural proteins are involved in the polyprotein processing, viral RNA synthesis and virus morphogenesis.
The HCV core protein is manly involved in the assembly of the nucleocapsid5. It consists of highly conserved 191 amino acids and mainly divided into the three major domains. Domain 1(1-117 amino acid) has basic amino acids which enhance the dimerization of viral RNA leading to the formation of nucleocapsid6. Domain 2 (118-174 amino acid) has less of the basic amino acids which are more hydrophobic in nature whereas Domain 3 (175-191 amino acid) has highly hydrophobic amino acids. Domain 2 and 3 are involved in the lipid transport and interaction with other HCV proteins namely E1, E2 and NS5A7.
The envelope protein E1 consists of C-terminal domain and is a transmembrane glycoprotein which regulates the membrane permeability changes and membrane association8. E2 is receptor binding protein which has 11 N-glycosylation sites. It is responsible for the viral particle attachment to the surface of the host cell. N –terminal of the E2 is referred as receptor binding region9. The p7 protein is hydrophobic transmembrane protein which forms the hydrophobic pores and allows the release of the viral particles to increase the infection by altering the membrane permeability. It is also involved in the late viral replication cycle10. Non-structural protein NS2 interacts with the E1, E2, p7 and other non-structural proteins to favor the viral assembly. It attracts the envelop proteins to the viral assembly site and promotes viral assembly11.
The N-terminal protease domain (NS3pro) of non-structural protein 3 (NS3) has role in proteolytic processing and the C-terminal region having RNA triphosphatase, RNA helicase and RNA-stimulated NTPase domain required for RNA replication. The serine protease domain of NS3 has major role in the replicative cycle of Flavivirus. The data shows that approximately 66% population of Northern India is found to be infected with NS3a12.
NS4A is involved to form the complex with the viral proteins such as NS3, NS4B and NS5A. it acts as a cofactor for proper functioning of the NS3 protein which results in increased enzymatic activities. It also helps in the viral replication at endoplasmic reticulum membrane by forming a complex with NS4B and NS5A13. NS4B interacts with NS5B to alter its polymerase activity and shows its significance in carcinogenesis. It is involved in the formation of the membranous structure which serves as a platform for the viral replication to happen14.
NS5A is proline rich phosphoprotein essential for the viral replication and assembly15. It is categorized into three domains. Domain 1is known as the zinc binding domain which forms the homodimer to get in contact with N- terminals. This domain is also involved in the binding of the RNA during the replication and may have role in switching between viral replication and translation. Domain 2 is responsible for the inhibition of the protein kinase PKR which is induced as a response to the IFN whereas domain 3 is poorly conserved [16]. NS5B is RNA dependent RNA polymerase which is responsible for the initiation of the replication cycle. Cooperation between the NS5B and p7 increase the Virion infectivity with the decrease in sphingomyelin level in Virion 17.
In-silico approaches have major role in HCV research from sequencing the genome to design the vaccine model before its implication in the wet lab. Our study is completely focused on reviewing bioinformatics tools and techniques used in the HCV research and to evaluate how bioinformatics has curved HCV research.
To find and analyze the bioinformatics tools and techniques used recently in the Hepatitis research various keywords and conditions were brought up in the action. After obtaining the query results the relevant research papers were reviewed and out of those related information was extracted. The extracted information were categorized into (1) Databases and tools, (2) Gene expression analysis and (3) Sequence alignment methods
For this entire work Pubmed database was used with 19 keywords, later on, all 19 queries (keywords) could be reduced to only 2 queries. Query 1: (hcv) OR hepatitis c) AND epitope -total hits were 1376 and Query 2: (anti hcv) OR anti hepatitis c) AND peptide -total hits were 3422.
Apart from bioinformatics tools and techniques used in HCV study, statistical methods used by the researchers were also mentioned. The frequency of above mentioned categories in different research papers was also calculated in order to determine which category was most frequently used by the researchers.
We in our study have categorized the information into Databases and tools, Gene expression analysis and Sequence alignment methods. We have discuss each part separately. The main objective behind the categorization was to put forward clear understanding of the information collected and it can be possible that different researchers will have different categorization methods. The tools which were used more frequently by the researchers were discussed along with the ones which were more crucial to carry out the research.
HCV Databases and Resources
Here, we will discuss the databases, resources and tools which were used by the researchers in HCV study. Most of the researchers who have followed in-silico approach used the databases in order to carry out their work meaningfully.
euHCVdb (European Hepatitis C Virus Database)
The database was developed by Combet18 and was available in 2006. By accessing the database the researchers can analyze the genetic and structural variability in the HCV sequences which can be implicated on vaccine and drug designing. The euHCVdb is internationally collaborated with US and Japan’s databases. In this database the researchers can access amino acid sequence of HCV proteins their 3D structures and functional analysis. The database is extended from the HCVDB (Hepatitis C virus database) which was developed in 199919. The euHCVdb is automatically annotated and updated every month from the EMBL (European Molecular Biology Laboratory) nucleotide sequence database. The database cab can be accessed by using the following web link: http://euhcvdb.ibcp.fr.
The Los Alamos hepatitis C sequence database
The database was developed by Kuiken et al20. At Los Alamos National Laboratory, US in 2004. The database is updated monthly from the HCV sequences present in Genbank. The database annotation includes sequence information in terms of genotype, subtype, and comparison to the reference HCV-H strain, sampling date, country, city and sampling tissue. Patient information provided in terms of age, gender, ALT level, HLA type, co-infection with HIV and hepatitis B, infection date, city, country, treatment and results. The web link to access the database is: http://hcv.lanl.gov.
HVDB (Hepatitis Virus Database)
This database was developed by Shin-I et al21. The database is a combined form of HCV, HBV and HEV databases. The database has around 44000 HCV (hepatitis C virus), 11000 HBV (hepatitis B virus) and 1600 HEV (Hepatitis E virus) sequences when it was made available to scientific community. The sequences present in the database were retrieved from INSDC (International Nucleotide Sequence Database Collaboration). The HCV master database contains the sequence sourced from DDBJ (DNA Databank of Japan). The reference map (loci, sub regions) is generated by comparing the retrieved sequence from DDBJ to the reference HCV genome. After that the sequence is placed under one of the three divisions which are C division, E1 division and NS5 division.
HCV pro
Kwofie et al22 developed the database which was available in 2011. The database provides the information about HCV protein-protein interactions. It contains the manually verified entries of Hepatitis virus-virus and virus-human protein interaction. The data was sourced from various literature and databases. The database provides extensive information on HCV proteins structure and function with development stage of drug and vaccine. In addition to this it provides the information of Hepatocellular Carcinoma genes which are coded into proteins and these proteins are linked to gene ontology, pathways, OMIM (Online Mendelian Inheritance in Man) and cross-referenced with various important annotations.
IEDB (Immune Epitope Database) resource
This resource has the various tools which are used to identify and analyze the epitopes. In a research this was employed to predict the MHC class 1 epitopes via ANN using the sequence method. The query sequence or the data are overlapped with the peptides & then their binding is checked by the prediction method selected for the analysis23.
ENCODE project database
Recently this database was utilized to create a bioinformatics pipeline. The database was used to obtain the raw form of the CHIP-sequence data. The obtained data was used to find the specific binding of the transcription factors to the alleles. The study was based on genetic prevention of the liver fibrosis (caused by HCV) by reducing the allele specific expression of the MERTK gene24.
Virus MINT database
This database stores the viral-human protein interaction information at the cellular level. As on 2008 this database had 5000 interactions out of which 490 were unique viral interactions from 110 viral strains. The database presents the query results in the form of graphical view. The database mainly contains the human protein interaction with viral proteins from the viruses such as Hepatitis C virus, human immunodeficiency virus 1, papilloma virus and other infectious and oncogenic viruses to human. The interactions stored were manually curated from MINT, IntAct and HIV-1 Human Protein Interaction Database25.
VirHostNet
This knowledge base is publicly available which contains the virus-virus, virus-host and host-host protein interaction information. It contains 2671 non-redundant virus-virus and virus-host interactions from different 180 viral species curated originally from the literatures and 10672 human protein interactions (68252 non-redundant entries) curated from publically available data26.
ViPR (Virus Pathogen Database and Analysis Resource)
This database stores the information of human pathogenic viruses’ family including flaviviridiae (HCV), other positive and negative-sense single stranded RNA viruses and double-stranded RNA virus. The main objective of the database is to provide single resource to access multiple virus research communities. ViPR contains the manually annotated information like sequences, epitopes, 3D protein structures obtained from GenBank, UniProt, PDB(Protein Data Bank), IEDB (Immune Epitope Database), PubMed and Gene Ontology Consortium27.
We used a variety of Databases for our search process, including the database search capabilities available under resources (Table 1).
Table (1):
Available Data resources for HCV research
Resource name |
Web Address |
Description |
Founded |
|---|---|---|---|
NCBI |
http://www.ncbi.nlm.nih.gov/. |
Biological Data bank |
1931 |
PDB |
http://www.rcsb.org/pdb/home/home.do |
Protein Structure Database |
1971 |
Pubmed |
http://www.ncbi.nlm.nih.gov/pubmed |
Literature database |
1996 |
Science Direct |
http://www.sciencedirect.com |
Literature database |
1997 |
Google |
http://www.google.com |
Search Engine |
1998 |
UniProt |
http://www.uniprot.org/ |
Protein Sequence Database |
2002 |
IEDB |
http://www.iedb.org/ |
Resource for immune epitopes |
2014 |
Important HCV Information Sources
In this part of information we have discussed some HCV information resources which basically provide the support to HCV infected individuals and spread awareness among the public including diagnosis and treatment information. Some of these organizations also offer a special training program for the HCV infected individuals.
Resource:
Hepatitis Foundation International
Address: http://www.hepatitisfoundation.org/
Scope: Hepatitis Patient Registry Network.
Audience: Patients, Health care professionals, General public
Description: The Hepatitis Foundation International (HFI) was established in 1944. It is a non-profit organization which aims to completely destroy viral hepatitis among the 550 million people around the world. In addition, it also provides the information regarding prevention of chronic liver disease, habits and practices which negatively effects liver.
Resource: HCV advocate
Address: http://hcvadvocate.org/
Scope: To provide correct information, support and make familiar with this information to the groups suffering from HCV, HIV/HCV co-infection including medical providers.
Audience: Patients, Health care professionals, General public
Description: It is a Hepatitis C support project, which is registered, non -funded organization established by Alan Franciscus in 1997. At present it is the reputed and well classified HCV publication in the U.S. Also offers training program to the HCV infected population.
Resource: Hep C
Address: http://hepc.liverfoundation.org/
Scope: Provide information regarding diagnosis, treatment and provide support the HCV infected individuals.
Audience: Patients, Health care professionals, General public
Description: Hepatitis C is American Liver Foundations online resource center which put forwards the information and support to the HCV positive population.
Resource: Centers for disease control and prevention
Address: http://www.cdc.gov/hepatitis/index.htm
Scope: HCV statistical, training information center
Audience: Patients, Health care professionals, General public
Description: Contains information about all the forms of hepatitis. All the statistical information, hepatitis outbreak information and training programs offered.
Resource: The Hepatitis Foundation of New Zealand
Address: http://www.hepatitisfoundation.org.nz/hepc
Scope: Hepatitis information center
Audience: Patients, Health care professionals, General Public
Description: Non -profit organization maintained by the Ministry of health, New Zealand. It look after for the people who are suffering from hepatitis B or C. Offers two courses, one “Hepatitis B FOLLOW up programme” and “Hepatitis C standard programme”. This foundation situated in New Zealand has performed important screening, vaccination and research programmes in New Zealand during their 30 years of work.
Resource: Public Health Agency of Canada
Address: http://www.phac-aspc.gc.ca/hepc/
Scope: HCV information center
Audience: Patients, Health care professionals, General Public
Description: The agency is maintained by the government of Canada to aware the people with various infectious diseases, their cause and treatment for reducing the frequency of infectious diseases, including the execution of the international research and development to the Canada’s people. Also used as a platform to exchange information of the Canada’s experts around the globe.
Tools used in HCV study
This protein-protein interaction network analysis was used in a study to obtain the protein-protein interaction information of HCV and human proteins. The purpose of using STRING was to form a network in which we can identify novel genes related to HCV and trace element metabolic process by preparing an identification approach28.
Tagident
This tool is used to determine the molecular weight and the isoelectric point of the unknown protein. Basis of determination is that it compares the query protein sequence with the sequences in the protein sequence database UniProtKB/Swissprot. By using this information the approximate location of the protein when it is analyzed under 2-Dimensional Gel Electrophoresis can be determined. In an HCV research this tool employed to identify the location of the proteins when they were running in the 2D Gel29.
Propred
This tool is for the prediction of the MHC epitopes. It contains the quantitative matrix which stores the scores which are generated by the experiments. Matrix compares the input peptide with that matrix which is generated by considering the properties of each amino acid and its location in an epitope. In a research these tools were extensively employed along with some other methods to determine the epitopes of the HCV, which are dominant in a group of the South African population on the basis of the binding score and other parameters which was presented by these tools as their output30.
BCPred
This tool is for the prediction of the B-cell epitopes. It uses the BCPred algorithm which takes the window size of 9 amino acid 75% specificity for the prediction. In a research this tool was employed to predict the epitopes and total 19 epitopes were predicted out of which 12 were antigenic as the antigenicity of the epitopes were confirmed by the vexijen version 2.0. This evaluation can be helpful in eliciting the desired immune response. For the T cell epitope prediction they used the online tool epijen which confirms the epitopic property. A total of 6 epitopes was determined by them using this31.
Clin Pro tools v2.0
This tool was utilized for the data analysis in the study focused on finding whether the serum proteome profiling is able to detect the treatment changes in the HCV-1b infected individuals. The uses of the tools include the normalization and recalibration of the spectra which was obtained from peptide profiling by MALDI-TOF/MS. The tool was utilized to statistically and visually analyze the data32.
M fold and I-TASSER
Both of these tools were used in a study to find out the 2D and 3D structure of the HCV-1a NS4A protein from Pakistan patients. The M fold web server was used to predict the folding of RNA sequence (2D structure) by obtaining single stranded frequency plot and energy dot plot for every RNA sequence33. I-TASSER was used to obtain the tertiary or 3D structure of the each amino acid sequence34.
SVM (Support Vector Machine) Model
In recent study machine learning methods like SVM were used to predict the interaction between the Hepatitis C virus protein and human protein. Cui’s SVM model showed average accuracy beyond 80% by using the feature number of times three consecutive amino acids present in a protein sequence. Emamjomeh’s SVM model using the features like amino acid composition, evolution information, PTM information, tissue information, pseudo amino acid composition and network centrality measures obtained accuracy of 83% when used on the human- HCV protein-protein interaction dataset (same dataset was used by the Cui)35.
IPA (Ingenuity Pathway Analysis)
This network analysis was performed in recent study to classify the proteins on the basis of their location inside the cell. In addition it also tells possible biological, molecular and biochemical functions of the protein36.
PatchDock
This program is used to perform the docking between the two molecules which can be protein, DNA, peptides and drugs. The process starts by converting the molecules into pieces called as patches on the basis of their surface shape and matching those patches with rest of the pieces by shape matching algorithm. The docking is performed in three steps named as Molecular shape representation, Surface patch matching and Filtering and Scoring 37,38. In study39 the drug vedroprevir docking was confirmed with active site of the HCV 1a NS3/4A protease.
The tools and databases which were used by the researchers during their HCV study are summarized in table 2. The databases have made the researchers to remain familiar with each and every up gradation in the HCV study. Numerous bioinformatics tools like Tagident which has allowed finding the PI value and molecular weight, then tools like Propred, BCPred helps in the determination of the epitopes of the HCV by using these tools they predicted the epitopes of the HCV. Algorithms like active paths algorithm which comes as a Cytoscape plug in also used which has made the researchers to find the network of the human and HCV protein interaction
Table (2):
Tools and the databases which were taken into consideration by the researchers during the course of HCV research
Year |
Resource Name |
Description |
Web Address |
|---|---|---|---|
2003 |
Pathway Studio |
Gene Expression |
http://www.ariadnegenomics.com/products/pathway-studio/ |
1994 |
ClustalW |
Multiple sequence alignment |
http://www.ebi.ac.uk/Tools/msa/clustalw2/ |
2001 |
ProPred |
Predict MHC Class-II binding regions in an antigen sequence |
http://www.imtech.res.in/raghava/propred/ |
2001 |
ProPred-1 |
Predict MHC Class-I binding regions in an antigen sequence |
http://www.imtech.res.in/raghava/propred1/ |
2003 |
Tagldent |
Identify isoelectric point and molecular weight |
http://web.expasy.org/tagident/ |
2008 |
BCPred |
Prediction of the B-cell epitopes |
http://ailab.ist.psu.edu/bcpred/ |
2011 |
HCVpro |
Hepatitis C virus protein interaction database |
http://cbrc.kaust.edu.sa/hcvpro/ |
2005 |
PatchDock |
For Docking Purpose |
http://bioinfo3d.cs.tau.ac.il/PatchDock/ |
Various bioinformatics techniques used in the HCV research, those techniques were broadly divided into four types namely considering sequence alignment, clustering/phylogeny, gene expression and databases or database searches studies along with the reference number of the paper in which these techniques were employed (Table 3). Out of all the reviewed papers the most frequently, the databases were mostly used by the researchers as they allow them to remain up to date with the current findings in the HCV study. Gene expression analysis allowed researchers to study the progression cycle of the HCV during different course of time.
Table (3):
Bioinformatics techniques used in HCV research
Reference |
Sequence Alignment |
Cluster/ Phylogeny Tree |
Gene Expression Analysis |
Database Searches / Database Assembly |
|---|---|---|---|---|
[18] |
– |
– |
– |
YES |
[19] |
– |
– |
– |
YES |
[31] |
– |
– |
– |
YES |
[40] |
YES |
– |
– |
YES |
[41] |
YES |
– |
– |
YES |
[43] |
– |
YES |
– |
YES |
[44] |
– |
– |
– |
YES |
[45] |
– |
YES |
– |
YES |
[46] |
– |
YES |
– |
– |
[47] |
YES |
– |
– |
– |
[48] |
– |
– |
YES |
YES |
[56] |
– |
YES |
– |
YES |
[57] |
– |
– |
– |
YES |
Total |
3 |
4 |
1 |
11 |
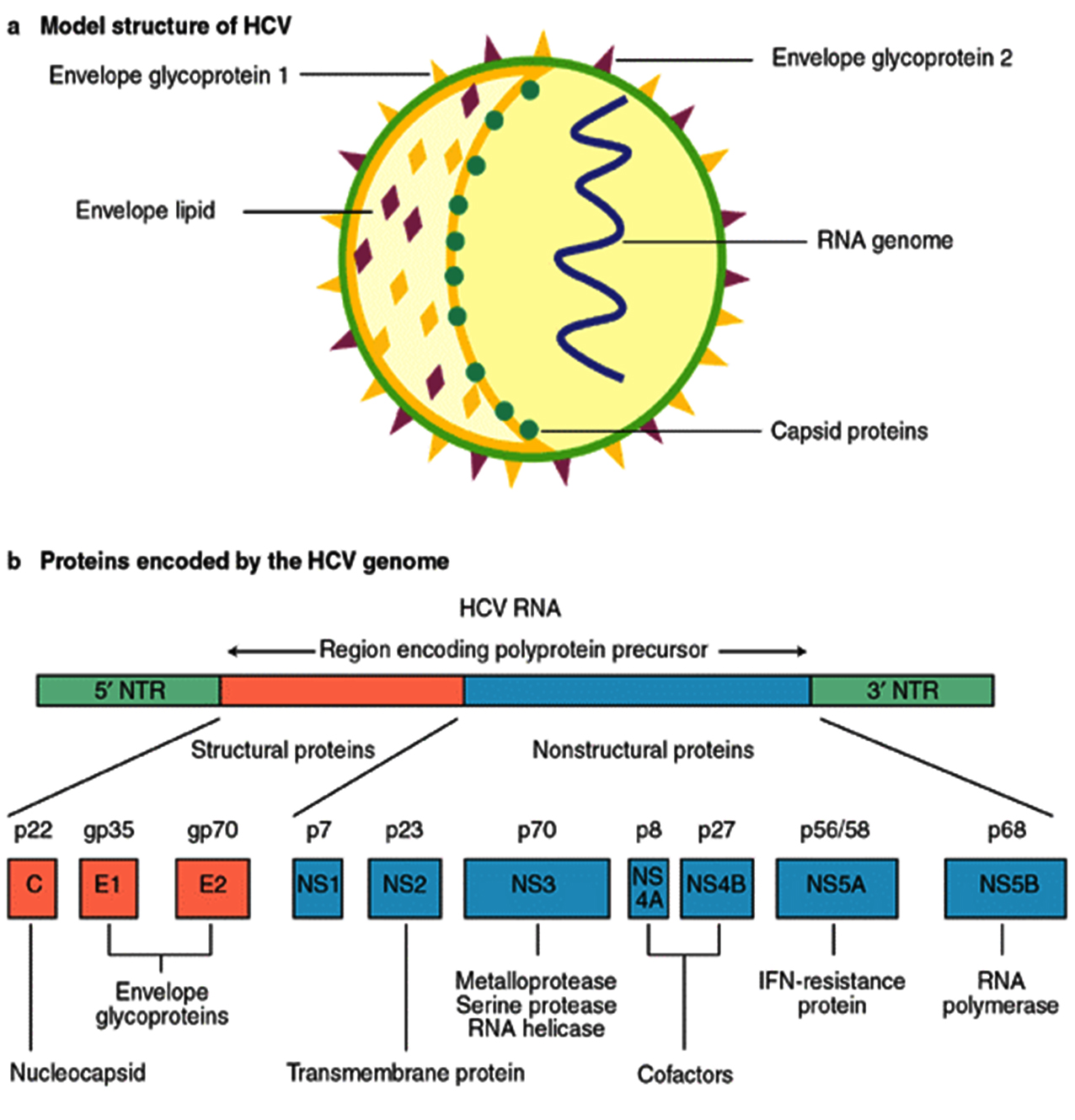
Fig. 1. Organization of the HCV proteins
The bioinformatics software employed mainly in the HCV research is mentioned in table 4. Out of all the work presented in the literature, none of the softwares were mentioned twice in different researches except the BLOSUM matrix, which was mentioned twice in40 and41. BLOSUM Matrix calculates and shows the no. of times a particular mutation taking place in a related protein family42. Table 5 highlights the different statistical tools used by the researchers in their HCV study. T test and equal variance were used in the form of software named as Minitab44.
Table (4):
Bioinformatics software used in HCV research
Program name |
Year |
No. of times mentioned |
Description |
|---|---|---|---|
Sequest |
1994 |
1 |
Mass spectroscopy oriented protein recognition tool |
Propred |
2001 |
1 |
MHC epitopes prediction |
Bimass |
2011 |
1 |
Calculation of peptide score |
Clustalv |
1992 |
1 |
Modified version of clustal for multiple sequence alignment |
Megalign |
1984 |
1 |
Pairwise & multiple sequence alignment of protein & DNA sequences |
Blosum |
1992 |
2 |
Substitution matrix for protein sequence alignment |
Jpred |
1998 |
1 |
Prediction of protein secondary structure |
Active path algorithm |
2005 |
1 |
Find the network or pathway active for a expression |
Rfam seed alignment |
2003 |
1 |
Multiple sequence alignment of RNA family from Rfam database |
Prosa |
1993 |
1 |
Error identification in 3D structure of protein |
Vaxijen |
2007 |
1 |
Antigen prediction server |
BCPred |
2008 |
1 |
B-cell epitope prediction |
Tagldent |
2003 |
1 |
Prediction of molecular weight & isoelectric point of unknown protein |
Table (5):
Statistical techniques used in HCV research
Statistical tool |
Year |
No. of times mentioned |
Description |
|---|---|---|---|
Z-score |
2008 |
1 |
Indicates how many standard deviations an element is from the mean. |
Ramachandran plot |
2013 |
1 |
A way to visualize energetically allowed regions |
Chi square test |
2012 |
2 |
Statistical based method to compare the observed data with practical data |
T-test |
2012 |
2 |
Statistical comparison of the means of the two data sets |
F-measure |
2014 |
1 |
Measure of accuracy by taking into consideration precision & recall value |
Gene Expression analysis
Gene expression analyses are performed to find out the level of expression of the different genes during the infection. Techniques like microarray and genetic linkage mapping were used. Study was done to find the expression of genes at different point of time during the HCV infection in a chimpanzee 48. The fold change in expression level of IFN-inducible genes were measured in which at day 7 the expression level was increased to 100 folds after this expression level was measured as normal in week 8.
Tools for gene expression analysis
MIDAS (Microarray Data Analysis System)
This software was developed by TM4 group. It is used to analyze the raw gene expression value from the spotfinder by doing the normalization (includes global, iterative linear regression and LOWESS normalization) and data analysis by t-Test and MAANOVA49.
SAM (Significance Analysis of Microarrays)
This tool is an excel add-on used for the analysis of various microarrays like Cdna or oligo array, SNP(Single Nucleotide Polymorphism) arrays, protein arrays etc. It relates the microarray expression data with clinical parameters including treatment, diagnosis categories, survival time etc50. In microarray gene expression analysis of HCV 3a genotype was done in early liver fibrosis and cirrhosis patients in which, MIDAS was used for normalization and SAM was used to identify significantly expressed genes during fibrosis stages51.
MAS (Microarray Analysis Suite)
This tool is used for the normalization and estimation of the microarray data for Affymetrix genechip. Normalization is done by the linear regression 52, 53.
RMA (Robust Multichip Average)
This software analyze the gene expression value for affymetrix genechip. The process is divided into background adjustment, quantile normalization and summarization54. MAS and MIDAS were used for microarray gene expression analysis in Hepatitis C virus and HCC (Hepatocellular Carcinoma) patients. RMA has better precision, consistency and specificity in detection of differential gene expression than MAS. MAS do reasonably good work on analyzing the brighter probe sets55.
Sequence Alignment Methods
Sequence alignment methods enable the researchers to find the homology between the DNA or amino acid sequences of different organism and find out the evolutionary relationship between them. In pairwise sequence alignment two sequences are compared with each other where as in multiple sequence alignment multiple sequences are compared with each other or with a single sequence. A group of researchers in 1994 published a paper in which they have sequenced the whole NS3b protein56. NS3b protein was the causative agent for HCV infection in North India. Pairwise and multiple sequence alignment tools were used by the researchers57. Abida’s research statement was brought to a conclusion that non-structural proteins of HCV virus control the activity of HCV virus inside the host and was significant to pathogenesis of hepatitis.
The research work of vikas helps us to understand human and viral protein interaction during the time of infection. By following the insilico approach with the help of partially identified transcripts of proteins they found the matches in Uniprot database, which enables them to design the epitopes 58. In their work we found the use of database Uniprot to obtain the sequences of the viral protein in short period of time. After various bioinformatics tools like propred and propred-1 used predict the epitopes from the protein sequences. The resulting epitopes were aligned with the multiple sequence alignment tool named as ClustalW. These bioinformatics tools were used to obtain the information of viral proteins at gene level.
The study of59 was focused to find the most pathogenic viral protein with the help of protein-protein interaction. Using the sequences obtained from the UniProt database, they predicted interaction of these protein sequences with Human proteins using HCVpro database.
Phylogenetic tree can simply referred to as evolutionary tree which shows evolutionary relationships among different biological species or molecules. Generally, the phylogenetic tree is based on similarities and variation in the physical or genetic features of organisms. The phylogenetic trees contain a lot of information about the inferred evolutionary relationships between a set of structural and non-structural proteins of hepatitis C virus (HCV).
In the figure 2, the horizontal dimension gives the amount of genetic change and it contains branches, the longer the branch in the horizontal dimension, the larger the amount of variation.
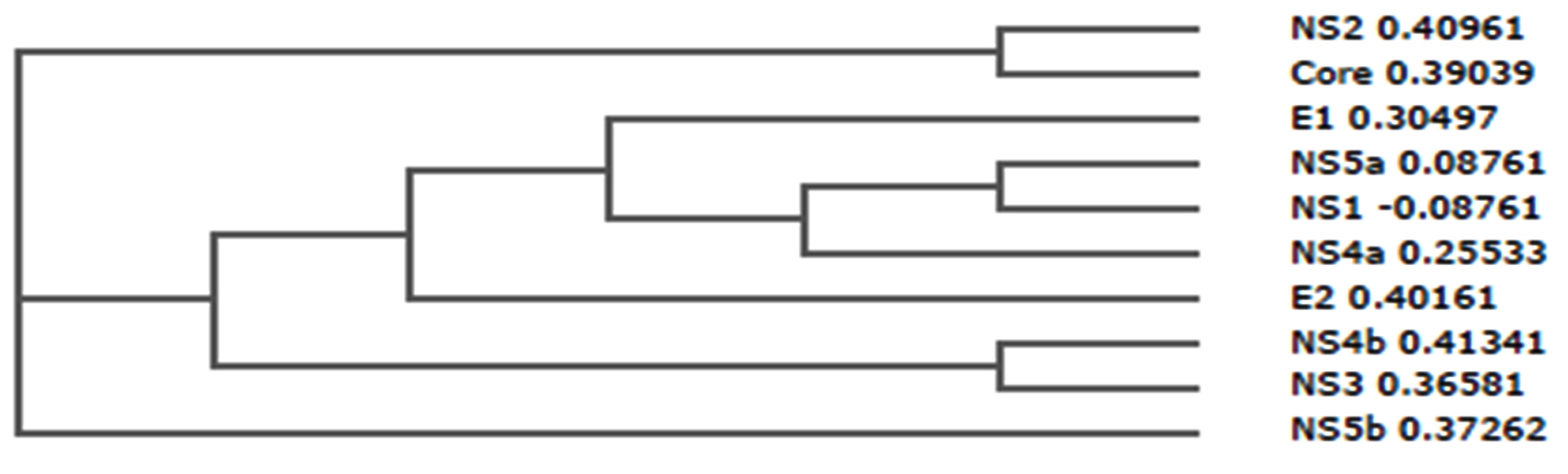
Fig. 2. Phylogenetic tree of structural and non-structural proteins of Hepatitis C virus (HCV)
From the result of comparative analysis of structural as well as non-structural protein shown in figure 2, NS2 (0.40961) and core (0.39039) were closely related, NS5a (0.08761) and NS1 (-0.08761) were closely related, NS4b (0.41341) and NS3 (0.36581) were also closely related.
However, E1 (0.30497), NS5a (0.08761), NS1 (-0.08761), NS4a (0.25533), E2 (0.40161), NS4b (0.41341), and NS3 (0.36581) were more related compare with NS2, core and NS5b which were deviated from the root of the tree.
Moreover, the strength of the similarities (relationships) is varied, for-instance, in the case of the seven proteins, the similarities between NS5a and NS1 is stronger than with NS4a, while that of NS4a is stronger than with E1, while that of E1 is stronger than with E2, while that of E2 is stronger than with NS4b and NS3.
urthermore, this evolutionary relationship is very significant in analysis of the T cell epitopes of hepatitis C virus due to the following reasons – (1) It enable us to predict what is the cause of immunogenicity of the virus; (2) It enable us to predict why some proteins have many epitopes compare with others and (3) It enable us to predict which features of amino acids should be consider more in design the model.
The present review discussed the work done to explore and analyze the use of the bioinformatics in the HCV study. The bioinformatics has played a major role from determining the sequence of the HCV proteins, pathway of interaction of those HCV proteins with the human proteins, the expression level of HCV infection during the different period of infection to the identification and the Insilco vaccine design against the HCV strains. Our research will help those who want to work in this field related to the HCV as a beginner and to those who are interested in working on the HCV choosing the bioinformatics as its field of study.
- Ku KS, Chodavarapu RK, Martin R, Miller MD, Mo H, Svarovskaia ES. Sequencing Analysis of NS3/4A, NS5A, and NS5B from Patients Infected with HCV Genotype 5 and 6. J. Clin. Microbiol. 2016;JCM-00238.
- Aly AM, Adel A, El-Gendy AO, Essam TM, Aziz RK. Gut microbiome alterations in patients with stage 4 hepatitis C. Gut Pathog. 2016; 8:42.
- Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, Abraham J, Adair T, Aggarwal R, Ahn SY, AlMazroa MA. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 2013; 380(9859):2095-128.
- Chen SL, Morgan TR. The natural history of hepatitis C virus (HCV) infection. Int J Med Sci. 2006; 3(2):47-52.
- Strosberg AD, Kota S, Takahashi V, Snyder JK, Mousseau G. Core as a novel viral target for hepatitis C drugs. Viruses 2010; 2(8):1734-51.
- Cristofari G, Ivanyi Nagy R, Gabus C, Boulant S, Lavergne JP, Penin F, Darlix JL. The hepatitis C virus Core protein is a potent nucleic acid chaperone that directs dimerization of the viral (+) strand RNA in vitro. Nucleic Acids Res. 2004; 32(8):2623-31.
- Boulant S, Vanbelle C, Ebel C, Penin F, Lavergne JP. Hepatitis C virus core protein is a dimeric alpha-helical protein exhibiting membrane protein features. J. Virol. 2005; 79(17):11353-65.
- Ciccaglione AR, Costantino A, Marcantonio C, Equestre M, Geraci A, Rapicetta M. Mutagenesis of hepatitis C virus E1 protein affects its membrane-permeabilizing activity. J. Gen. Virol. 2001; 82(9):2243-50.
- Fraser J, Boo I, Poumbourios P, Drummer HE. Hepatitis C virus (HCV) envelope glycoproteins E1 and E2 contain reduced cysteine residues essential for virus entry. J. Biol. Chem. 2011; 286(37):31984-92.
- Steinmann E, Penin F, Kallis S, Patel AH, Bartenschlager R, Pietschmann T. Hepatitis C virus p7 protein is crucial for assembly and release of infectious virions. PLoS Pathog. 2007; 3(7):e103.
- Popescu CI, Callens N, Trinel D, Roingeard P, Moradpour D, Descamps V, Duverlie G, Penin F, Héliot L, Rouillé Y, Dubuisson J. NS2 protein of hepatitis C virus interacts with structural and non-structural proteins towards virus assembly. PLoS Pathog. 2011; 7(2):e1001278.
- Singh S, Malhotra V, Sarin S. Distribution of hepatitis C virus genotypes in patients with chronic Hepatitis C infection in India. Ind. J. Med. Res. 2004; 119:145-148.
- Nomura-Takigawa Y, Nagano-Fujii M, Deng L, Kitazawa S, Ishido S, Sada K, Hotta H. Non-structural protein 4A of Hepatitis C virus accumulates on mitochondria and renders the cells prone to undergoing mitochondria-mediated apoptosis. J. Gen. Virol. 2006; 87(7):1935-45.
- Einav S, Gerber D, Bryson PD, Sklan EH, Elazar M, Maerkl SJ, Glenn JS, Quake SR. Discovery of a hepatitis C target and its pharmacological inhibitors by microfluidic affinity analysis. Nature Biotechnol. 2008; 26(9):1019-27.
- Huang Y, Staschke K, De Francesco R, Tan SL. Phosphorylation of hepatitis C virus NS5A nonstructural protein: a new paradigm for phosphorylation-dependent viral RNA replication ? Virology 2007; 364(1):1-9.
- Love RA, Brodsky O, Hickey MJ, Wells PA, Cronin CN. Crystal structure of a novel dimeric form of NS5A domain I protein from hepatitis C virus. J, Virol. 2009; 83(9):4395-403.
- Aligeti M, Roder A, Horner SM. Cooperation between the hepatitis C virus p7 and NS5B proteins enhances virion infectivity. J. Virol. 2015; 89(22):11523-33.
- Combet C, Garnier N, Charavay C, Grando D, Crisan D, Lopez J, Dehne-Garcia A, Geourjon C, Bettler E, Hulo C, Le Mercier P. euHCVdb: the European hepatitis C virus database. Nucleic Acids Res. 2007; 35(suppl 1):D363-6.
- Combet C, Penin F, Geourjon C, Deleage G. HCVDB: hepatitis C virus sequence database. Appl. Bioinformatics 2004; 3(4):237-240.
- Kuiken C, Yusim K, Boykin L, Richardson R. The Los Alamos hepatitis C sequence database. Bioinformatics 2005; 21(3):379-84.
- Shin I T, Tanaka Y, Tateno Y, Mizokami M. Development and public release of a comprehensive hepatitis virus database. Hepatol Res. 2008; 38(3):234-43.
- Kwofie SK, Schaefer U, Sundararajan VS, Bajic VB, Christoffels A. HCVpro: hepatitis C virus protein interaction database. Infect. Genet. Evol. 2011; 11(8):1971-7.
- Zhang Q, Wang P, Kim Y, Haste-Andersen P, Beaver J, Bourne PE, Bui HH, Buus S, Frankild S, Greenbaum J, Lund O. Immune epitope database analysis resource (IEDB-AR). Nucleic Acids Res. 2008; 36(suppl 2):W513-8.
- Cavalli M, Pan G, Nord H, Wallén Arzt E, Wallerman O, Wadelius C. Genetic prevention of HCV induced liver fibrosis by allele specific down regulation of MERTK. Hepatol. Res. 2016;doi: 10.1111/hepr.12810.
- Chatr-Aryamontri A, Ceol A, Peluso D, Nardozza A, Panni S, Sacco F, Tinti M, Smolyar A, Castagnoli L, Vidal M, Cusick ME. VirusMINT: a viral protein interaction database. Nucleic Acids Res. 2009; 37(suppl 1):D669-73.
- Guirimand T, Delmotte S, Navratil V. VirHostNet 2.0: surfing on the web of virus/host molecular interactions data. Nucleic Acids Res. 2015; 43(D1):D583-7.
- Pickett BE, Greer DS, Zhang Y, Stewart L, Zhou L, Sun G, Gu Z, Kumar S, Zaremba S, Larsen CN, Jen W. Virus pathogen database and analysis resource (ViPR): A comprehensive bioinformatics database and analysis resource for the coronavirus research community. Viruses 2012; 4(11):3209-26.
- Zhu L, Chen X, Kong X, Cai YD. Investigation of the roles of trace elements during hepatitis C virus infection using protein-protein interactions and a shortest path algorithm. Biochimica et Biophysica Acta (BBA)-General Subjects 2016; 1860(11):2756–2768.
- Garcia AS, Ibarra CPR, Sanchez ARR, Lopez RO, Juarez AG, Chavez JM, Rodriguez HM, Estilla AMR. Use of proteomic analysis tools to identify HCV-proteins down-regulated by acetylsalicylic acid. Ann. Hepatol. 2013; 12(5):725-732.
- Sing NP, Puren AJ, Bowyer SM. Sequence based in-silico analysis of well studied Hepatitis C Virus epitopes and their variants in other genotypes (particularly genotype 5a) against South African human leukocyte antigen backgrounds. BMC Immunol. 2012; 13:67.
- Idrees S, Ashfaq UA. Structural analysis and epitope prediction of HCV E1 protein isolated in Pakistan: an in-silico approach. Virol. J. 2013; 10(1):1.
- Yen YH, Wang JC, Hung CH, Lu SN, Wang JH, Hu TH, Kee KM, Hsiao CC, Lee CM. Serum proteome predicts virological response in chronic hepatitis C genotype 1b patients treated with pegylated interferon plus ribavirin. J. Formos. Med. Assoc. 2015; 114(7):652-8.
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003; 31(13):3406-15.
- Hussain A, Idrees M, Asif M, Ali L, Rasool M. Phylogenetic and 2D/3D Analysis of HCV 1a NS4A Gene/Protein in Pakistani Isolates. Hepat. Mon. 2015; 15(6):e19936.
- Kim B, Alguwaizani S, Zhou X, Huang DS, Park B, Han K An improved method for predicting interactions between virus and human proteins. J. Bioinform. Comput. Biol. 2016; 1650024.
- Ye F, Xin Z, Han W, Fan J, Yin B, Wu S, Yang W, Yuan J, Qiang B, Sun W, Peng X. Quantitative Proteomics Analysis of the Hepatitis C Virus Replicon High-Permissive and Low-Permissive Cell Lines. PLoS One 2015; 10(11):e0142082.
- Duhovny D, Nussinov R, Wolfson HJ. Efficient unbound docking of rigid molecules. InInternational Workshop on Algorithms in Bioinformatics. Springer Berlin Heidelberg. 2002; 185-200.
- Schneidman-Duhovny D, Inbar Y, Nussinov R, Wolfson HJ. PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res. 2005; 33(suppl 2):W363-7.
- Palanisamy N, Lennerstrand J. Biophysical Studies on HCV 1a NS3/4A Protease and Its Catalytic Triad in Wild Type and Mutants by the In Silico Approach. Interdisciplinary Sciences: Computational Life Sciences 2016; 16:1-4.
- Walewski JL, Gutierrez JA, Elliman WB, Stump DD, Keller TR, Rodriguez A, Benson G, Branch AD. Mutation master: profiles of substitutions in hepatitis c virus RNA of the core, alternate reading frame, and NS2 coding regions. Rna 2002; 8(5):557-571.
- Tellinghuisen TL, Marcotrigiano J, Gorbalenya AE, Rice CM. The NS5A protein of hepatitis C virus is a zinc metalloprotein. J. Biol. Chem. 2004; 279(47):48576-48587.
- S. Henikoff, J.G. Henikoff, Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. 1992; 89:10915-10919.
- Reiss DJ, Avila-Campillo I, Thorsson V, Schwikowski B, Galitski T. Tools enabling the elucidation of molecular pathways active in human disease: application to Hepatitis C virus infection. BMC Bioinform. 2005; 6(1):154.
- Yamasaki LH, Arcuri HA, Jardim AC, Bittar C, de Carvalho-Mello IM, Rahal P. New insights regarding HCV-NS5A structure/function and indication of genotypic differences. Virol. J. 2012; 9(1):1.
- Leung P, Bull R, Lloyd A, Luciani F. A bioinformatics pipeline for the analyses of viral escape dynamics and host immune responses during an infection. BioMed Res. Int. 2014;2014.
- KayvanJoo AH, Ebrahimi M, Haqshenas G. Prediction of Hepatitis C virus interferon/ribavirin therapy outcome based on viral nucleotide attributes using machine learning algorithms. BMC Res Notes 2014; 7:565.
- Verbist BM, Thys K, Reumers J, Wetzels Y, Van der Borght K, Talloen W, Aerssens J, Clement L, Thas O. VirVarSeq: a low-frequency virus variant detection pipeline for Illumina sequencing using adaptive base-calling accuracy filtering. Bioinformatics 2015; 31(1):94-101.
- Bigger CB, Brasky KM, Lanford RE. DNA microarray analysis of chimpanzee liver during acute resolving hepatitis C virus infection. J. Virol. 2001; 75(15):7059-7066.
- Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N, Braisted J, Klapa M, Currier T, Thiagarajan M, Sturn A, Snuffin M, Rezantsev A, Popov D, Ryltsov A, Kostukovich E, Borisovsky I, Liu Z, Vinsavich A, Trush V, Quackenbush J. TM4: a free, open-source system for microarray data management and analysis. Biotechniques 2003; 34(2):374-8.
- Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. 2001; 98(9):5116-21.
- Ahmad W, Ijaz B, Hassan S. Gene expression profiling of HCV genotype 3a initial liver fibrosis and cirrhosis patients using microarray. J. Transl. Med. 2012; 10(1):1.
- Liu WM, Mei R, Di X, et al. Analysis of high density expression microarrays with signed-rank call algorithms. Bioinformatics2002; 18:1593-9.
- Li C, Wong WH. Model-based analysis of oligonucleotide arrays: model validation, design issues and standard error applications. Genome Biol. 2001; 2:1-11.
- Li C, Wong WH. DNA-chip analyzer (dchip). In: Parmigiani G, Garrett ES, Irizarry RA, Zeger SL, Eds. The Analysis of Gene Expression Data: Methods and Software. New York: Springer-Verlag 2003;120–41.
- Mas VR, Maluf DG, Archer KJ, Yanek K, Williams B, Fisher RA. Differentially expressed genes between early and advanced hepatocellular carcinoma (HCC) as a potential tool for selecting liver transplant recipients. Molecular Medicine-Cambridge Ma Then New York-2006; 12(4/6):97.
- Chayama K, Tsubota A, Koida I, Arase Y, Saitoh S, Ikeda K, Kumada H. Nucleotide sequence of Hepatitis C virus (type 3b) isolated from a Japanese patient with chronic hepatitis C. J. Gen. Virol. 1994; 75:3623-3628.
- Shehzadi, Rhman S, Idrees M. Promiscuous prediction and conservancy analysis of CTL binding epitopes of HCV 3a viral proteome from Punjab Pakistan: an in Silico Approach. Virol. J. 2011; 8:55.
- Kaushik V, Singh J, Kaur R, Kumar A. In silico peptide based vaccine design against non- structural protein 3 of Hepatitis C virus, Wilkes100 – ICCS 2013 2013;498-503.
- Kaushik V, Chauhan G, Singh J. In Silico peptide based vaccine design against non-structural Protein 5 of Hepatitis C Virus. Int. J. Pharm. Pharmaceut. Sci. 2014; 6(9):80-82.
© The Author(s) 2017. Open Access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License which permits unrestricted use, sharing, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.