


ISSN: 0973-7510
E-ISSN: 2581-690X
The ongoing coronavirus disease 2019 (COVID-19) pandemic, putatively caused by the widespread transmission of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has resulted in significant mortality worldwide. The highly varied epidemiology of the disease both temporally and geographically has garnered much attention. The present study aimed to gain a deeper understanding of the varied geospatial disease epidemiology during the first wave of the pandemic. The highly mutable spike (S) protein, which confers fitness to SARS-CoV-2 for its survival and spread was studied using representative sequences determined from the initial phase of the pandemic. Adaptive evolution and selection pressure analysis of 311 whole-genome sequences from across the world including Asia (n=105), Europe (n=101), and the United States (n=105) was performed. A high selection pressure at position 614 of the S protein with a dN/dS (non-synonymous/synonymous substitutions per site) ratio of 124.3 for Asia and 867.9 was predicted for Europe. This positively selected site (i.e. 614) was located in the S1 domain (amino acids 14-680), which acts in binding to the angiotensin-converting co-enzyme 2 (ACE2) receptor. The US strains did not exhibit significant positive selection at position 614. In addition, 10 sites (144, 241, 255, 262, 263, 276, 439,517, 528, and 557) in domain 1 and 19 sites (692, 709, 723, 752, 862, 864, 877, 892, 939, 951, 1015, 1060, 1076, 1114, 1116, 1128, 1176, 1235 and 1240) in domain 2 of the S protein mediating viral entry into host cells, exhibited significant negative selection among European strains of (SARS-CoV-2), however, no negative selection was observed in the Asian and US groups. The D614G spike protein variant has been correlated with fatal outcomes in European population and countries including Italy, France, Belgium, and Spain. D614G variants under high selective pressure in the Asian and European strains were also observed. In addition, the presence of 29 negatively selected codon sites under low selection pressure in the European group may imply improved viral fitness compared with strains circulating in other continents. In conclusion, selective pressure on the S protein, with maximum substitution rate, may have facilitated adaptive evolution of the virus and contributed to the worldwide spread of the virus.
COVID-19, D614G, Evolution, Positive pressure, SARS-CoV-2, Spike protein
The emergence of the novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in December 2019 in China, with its rapid international spread, has led to a global health emergency. By February 27, 2021, >113 million cases were confirmed with >2.5 million deaths attributed to coronavirus disease 2019 (COVID-19) according to the World Health Organization.1 The virus has spread globally, resulting in the ongoing COVID-19 pandemic. The epidemiology of the disease has been highly varied and region- and time-specific. During the first wave of the pandemic, before June 2020, the United States (US) had borne the greatest disease burden with > 9.4 million cases and a mortality rate of 713 deaths per million population. On the other hand, Europe (~4 million cases) reported the highest mortality rate (11,793 deaths/million). Although the total number of cases in Asia exceeded 7 million the mortality rate remained low at 199 deaths per million in Southeast Asia. The difference in disease burden and outcome across geographically distinct regions could have been due to either host-related factors or the circulating viral strain and its adaptation to the geographical locations and the population. The first whole-genome sequence was published on January 5, 2020, and thousands of genomes have been sequenced since then. The present investigation studied differences among circulating strains and the adaptive evolutionary pressures on the SARS-CoV-2 across various geographical regions during the first wave of the pandemic using sequencing data obtained between January and June 2020. The 30 kilobase-pair genome of the SARS-CoV-2 codes for the envelope (E), membrane (M), nucleocapsid (N), and spike (S) proteins. The S protein exhibits the greatest variability and mutational rates; therefore, it is thus important to characterize this protein to delineate global and region-specific evolutionary trends.
A total of 311 whole-genome sequences were retrieved from the GISAID server (https://www.gisaid.org/) across the world. Filters were applied to retrieve sequencing data from the first wave of the pandemic (January to June 2020) for three distinct geospatial regions. The downloaded sequences were grouped geographically into Asia (group I [n=105], Europe (group II [n=101], and the US (group III, [n=105]) (Fig. 1). After deletion of duplicated sequences, 40 from group I, 35 from group II, and 28 from group III were used for analysis. The complementary DNA sequences of 3822 nucleotides were truncated at nucleotide positions 21,563 to 25,384 nucleotide positions. The selected nucleotide sequences of each dataset were aligned using ClustalW (https://www.genome.jp/tools-bin/clustalw) and were further analyzed using HyPhy software under the data-monkey web server (https://www.datamonkey.org/). All 1273 codon sites of the S protein were analyzed as dN/dS ratio (non-synonymous/synonymous substitutions per site) which was calculated using the IFEL approach (http://classic.datamonkey.org/help/fel.php). For the dN/dS ratio per site, p<0.1 was considered to be statistically significant. Positively and negatively selected sites were mapped onto the crystallographic structure of the SARS-CoV-2 surface S protein using PyMol (https://pymol.org/2/) for spatial depiction of the selected codons.
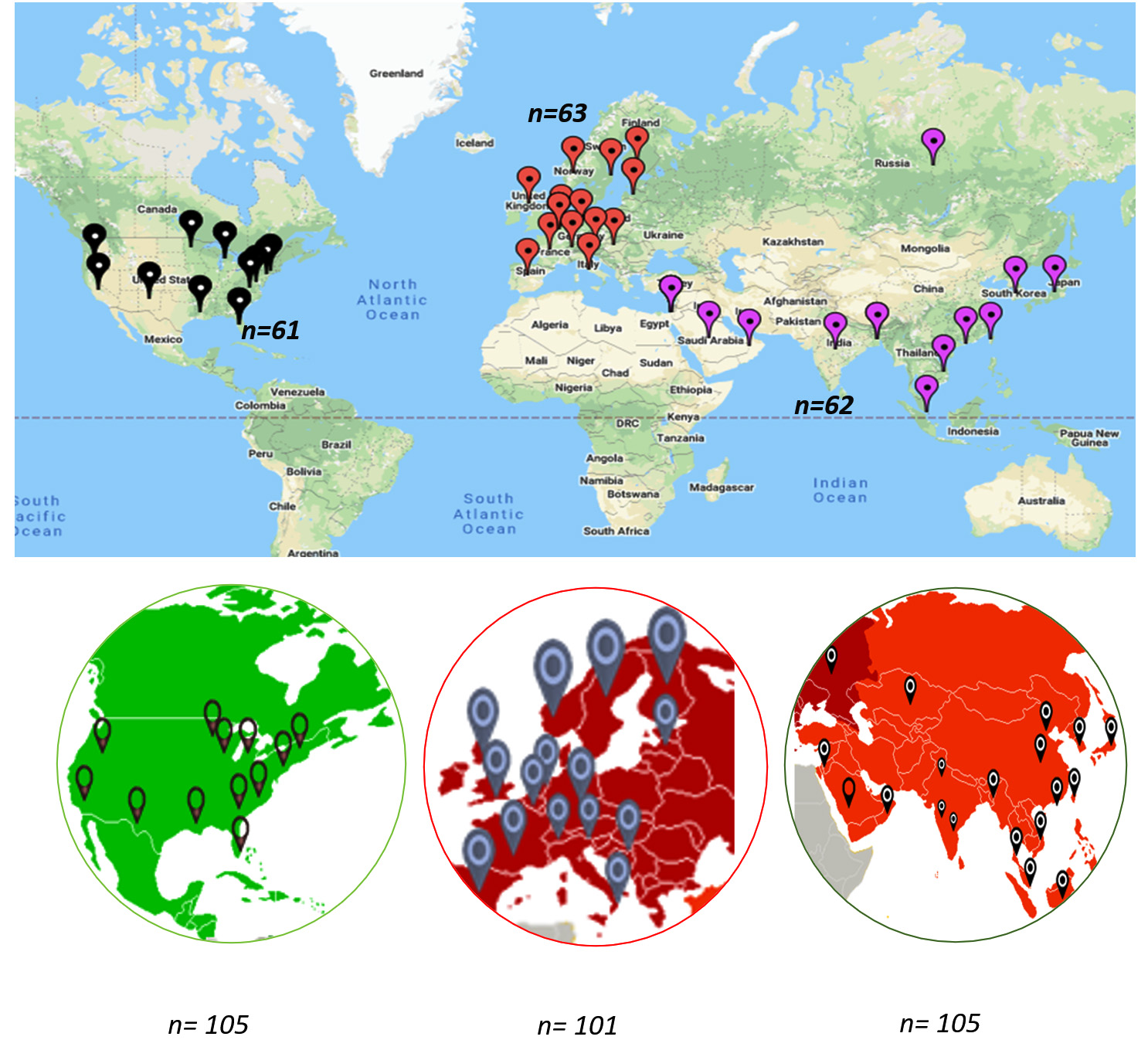
Fig. 1. World map representing Geospatial dominance of D614G mutations showing for the USA in Black (61/105), Europe in Red (63/101), and Asia in purple (62/105).
Selection pressure analysis of the Asia, Europe, and the US strains exhibited a high dN/dS ratio at codon position 614 of the S protein in the European and Asian strains, indicating positive selection (Fig. 2). The dN/dS ratio at codon 614 was higher in European strains (dN/dS: 867.9; p= 0.023) than in the Asian strains (dN/dS: 124.3; p= 0.019); however the US strains did not exhibit any significance in positive selection at the site. On mapping of the positively and negatively selected codon sequences on the three-dimensional crystallographic structure of the S protein (6XS6, D614G [aspartate to glycine] variant), residue 614 was found to be located in the S1 domain (Fig. 3).

Fig. 2. Graphical representation of positive and negative selection pressure sites. The single upward peak represents positive selection at position 614 whereas other downwards pointing peaks represent negative selection at different sites.
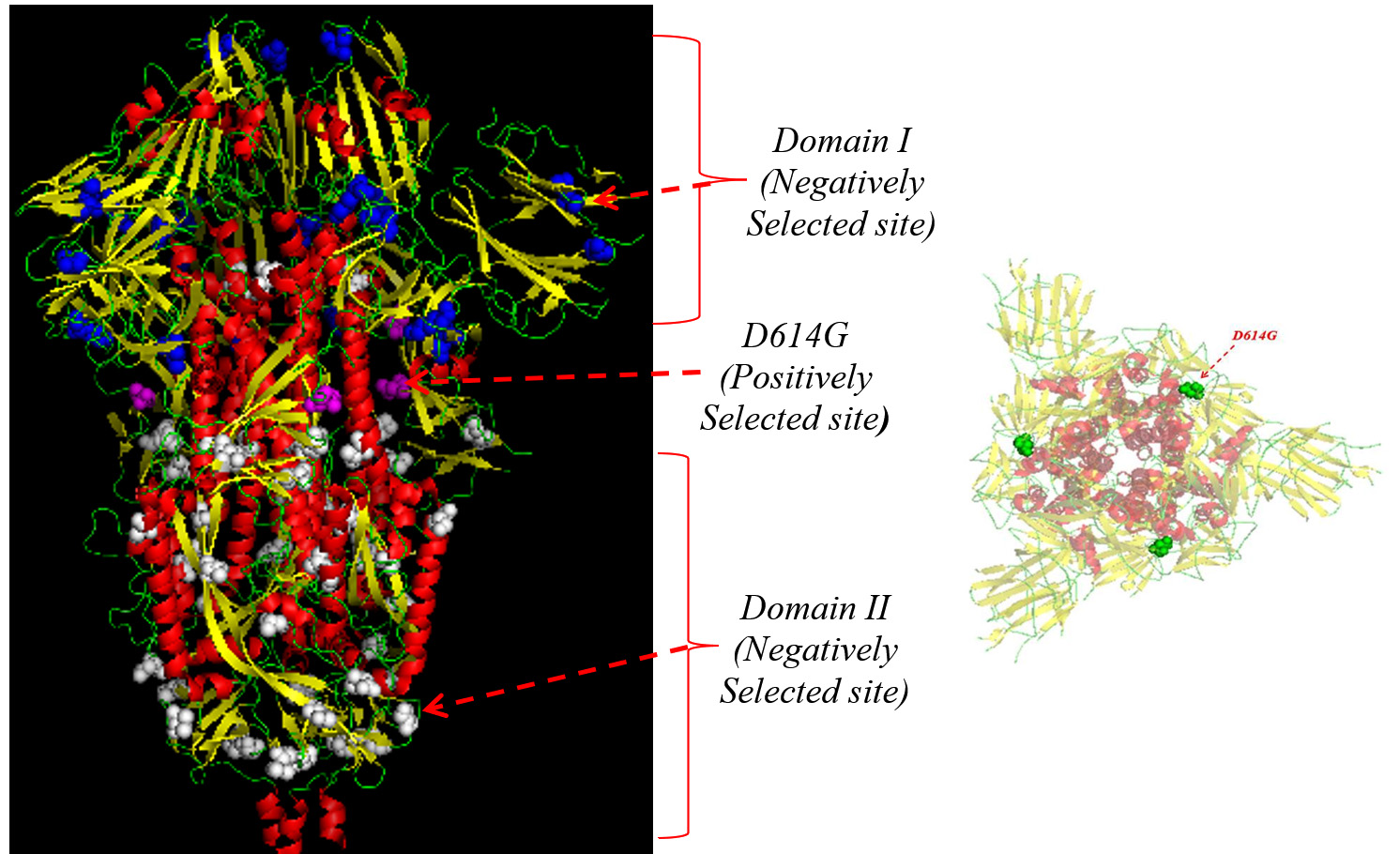
Fig. 3. Trimeric spike protein structure showing D614G sites (purple) as a positively selected site, negatively selected sites in the Domain-I (blue), and Domain-II (white) in the side view. In the top view representation of the structure, positively selected sites were presented in green.
Evidence of negative selection was observed only in the European strains, with 29 negatively selected sites distributed across the S protein without being specifically restricted to a particular region. On mapping the sites on the crystallographic structure, 10 sites (144, 241, 255, 262, 263, 276, 439,517, 528, and 557) were located in domain 1 and 19 sites (692, 709, 723, 752, 862, 864, 877, 892, 939, 951, 1015, 1060, 1076, 1114, 1116, 1128, 1176, 1235, and 1240) were located in domain 2 of the S protein (Fig. 3) with no significant negatively selected sites in the Asian and US strains. Thus, a total of 186 D614G variants were observed on mutational analysis using MEGA 7.0 software among the strains data downloaded from various geographical locations (n=311) (Fig. 1).
Table (1):
Geographical distribution of D614 and D614G substitution in USA, Europe and Asia.
| Country/ States |
Total Sequences |
D 614 | G 614 | % D614G |
|---|---|---|---|---|
| United States of America | ||||
| California | 21 | 10 | 11 | 52.3 |
| New-York | 9 | 0 | 9 | 100 |
| Washington | 23 | 13 | 8 | 34.7 |
| Oregon | 9 | 2 | 7 | 77.7 |
| Cruise | 7 | 7 | 0 | – |
| Virginia | 5 | 0 | 5 | 100 |
| Wisconsin | 8 | 4 | 4 | 50 |
| Connecticut | 4 | 0 | 4 | 100 |
| Louisiana la | 4 | 0 | 4 | 100 |
| Minnesota mn | 6 | 3 | 3 | 50 |
| Michigan | 2 | 0 | 2 | 100 |
| New Mexico | 1 | 0 | 1 | 100 |
| 105 | 44 | 61 | ||
| Asia | ||||
| Russia | 13 | 0 | 13 | 100 |
| India | 17 | 8 | 9 | 52.9 |
| Bangladesh | 9 | 0 | 9 | 100 |
| Japan | 8 | 0 | 8 | 100 |
| Hong Kong | 9 | 1 | 8 | 88.8 |
| Israel | 4 | 0 | 4 | 100 |
| Vietnam | 3 | 0 | 3 | 100 |
| Singapore | 18 | 16 | 2 | 11.1 |
| South Korea | 8 | 6 | 2 | 25 |
| Saudi Arabia | 3 | 1 | 2 | 66.6 |
| Oman | 1 | 0 | 1 | 100 |
| Taiwan | 1 | 0 | 1 | 100 |
| China | 5 | 5 | 0 | 0 |
| Kazakhstan | 3 | 3 | 0 | 0 |
| Malaysia | 3 | 3 | 0 | 0 |
| 105 | 43 | 62 | ||
| Europe | ||||
| Italy | 20 | 9 | 11 | 55 |
| Switzerland | 11 | 1 | 10 | 90.9 |
| England | 13 | 5 | 8 | 61.5 |
| Latvia | 5 | 0 | 5 | 100 |
| France | 8 | 4 | 4 | 50 |
| Finland | 6 | 2 | 4 | 66.6 |
| Cz. Republic | 5 | 1 | 4 | 80 |
| Austria | 3 | 0 | 3 | 100 |
| Germany | 4 | 1 | 3 | 75 |
| Spain | 7 | 4 | 3 | 42.8 |
| Belgium | 3 | 1 | 2 | 66.6 |
| Sweden | 7 | 5 | 2 | 28.5 |
| Netherlands | 3 | 1 | 2 | 66.6 |
| Norway | 1 | 0 | 1 | 100 |
| Hungary | 1 | 0 | 1 | 100 |
| Scotland | 1 | 1 | 0 | – |
| Wales | 3 | 3 | 0 | – |
| 101 | 38 | 63 | ||
Since the emergence of SARS-CoV-2, it has been evolving. Its high transmissibility, persistence in the community, viral shedding through various bodily secretions including nasal and throat secretions (sputum and saliva), and stool has made it more adaptable. Moreover, the rapid adaptation to human-to-human transmission with increasing morbidity and mortality in patients with underlying comorbidities has generated a public health concern.
Disease behavior has been highly varied and unpredictable among various population groups. While reports from European countries initially demonstrated the highest mortality per million population, the US reported the highest number of cases during the first wave. Asian countries on the other hand have witnessed the lowest mortality rate per million. The difference in disease behavior could be attributed to differences in the host population or circulating viral strain. The highly variable ‘S’ glycoprotein of the SARS-CoV-2 which facilitates viral entry into the host cell plays a pivotal role in determining the transmissibility and infectivity of the virus, therefore studied to understand any adaptive evolutionary changes. The presence of a positively selected site (D614G) in the Asian and European groups points to diversifying selection in the European and Asian groups. The presence of diversifying selection in the S1 domain of the S protein could be immunologically driven or could imply viral adaptation to the new human host. Positively selected sites (diversifying selection) were also observed in the initial phase of the SARS epidemic in 2002 when the virus crossed a species barrier to adapt to the human host.
Numerous comparisons have been made between the present pandemic and the previous H1N1 and SARS outbreaks. Both infections were caused by novel RNA viruses that crossed the species barrier to infect the respiratory tract causing respiratory symptoms in humans. The HA and spike proteins of the H1N1 and SARS viruses respectively plays a part in viral attachment, which is homologous to the function of the S protein in the SARS-CoV-2. Adaptive evolutionary studies investigating Inf A (H1N1) viruses from the early and post-pandemic period reported positively selected residues located on the conserved HA stem region and HA globular head respectively, possibly due to adaptation to the new host and/or may be attributed to host immune response. Positions of positively selected sites on mushroom-shaped HA proteins in Inf A (H1N1) was markedly different between the pandemic and post-pandemic periods.2
In contrast, the location of positively selected sites in the variable head portion of the SARS-CoV-2 S protein during the present phase of the pandemic could suggest a diversifying selection that could be driven by the host immune response. Other studies investigating selection pressure have analyzed orf1ab genes and S proteins from a dataset of 91 and 39 SARS-CoV-2 sequences respectively. Most sites for both genes demonstrated ω values of <1, indicating purifying selection, with 7 sites under negative selection (215, 474, 541, 809, 820, 921, and 1044), and none under positive selection.
In the SARS epidemic of 2002, the S protein played a key role in the virus crossing the species barrier and was therefore under high selective pressure. Selection pressure studies during the SARS epidemic in the early and late epidemic phases have reported several positively selected sites in the early and mid-phases of the pandemic implying viral adaptation to the new human host with the majority of positively selected sites located in the S1 domain of the S protein responsible for binding to human angiotensin-converting enzyme-2 (ACE2) receptors. The late phase of the epidemic, however, provided evidence of purifying selection with no positively selected sites reported.
Genomic analysis of 103 SARS-CoV-2 genome sequences suggested 96.2% similarity to bats infected with the SARS-related coronavirus (SARSr-CoV; RaTG13) as reported in earlier studies from China. Variability among SARS-CoV-2 and SARSr-CoV; RaTG13 in genomic nucleotides (4%) and at neutral sites (17%), suggests a degree of divergence between them.3 Additionally, the L lineage (more prevalent, [approximately 70%]) and S lineage (approximately 30%) of SARS-CoV-2 were also found. Tang et al.,4 highlighted the role of natural selection leading to new variations in the receptor-binding domain (RBD) of the S protein of SARS-CoV-2 and pangolin SARSr-CoVs.
Zhang et al.,5 identified positively selected amino acid sites on the surface of the S protein (239, 311, 479, 609, 743, 778, 1148, and 1163), thereby suggesting its crucial role in SARS-CoV transmission and survival. The variation in positive selective sites and pressures have in large part been identified in the RBD and HR1–HR2 regions of the S protein and tend to be changeable in different epidemics before stabilization. By influencing receptor recognition and/or membrane fusion, such variations help in understanding the molecular adaptation of the S protein for interspecies transmission from animals to humans.
The S protein is an important determinant of the pathogenicity of SARS-CoV-2 and is highly susceptible to a large number of mutations.6 Laamarti et al.,7 analyzed the genetic variations in 3067 whole genomes of SARS-CoV-2 collected from 55 countries and found 512 variant sites with a non-synonymous effect in addition to 10 hotspot mutations distributed in six SARS-CoV-2 genes. Among genomes from the US isolates, V483A mutation in the S receptor was also identified, suggesting the specificity of certain genotypes to the geographical location. Consistent with the present study, Ou et al.8 identified eight stains from China, the US, and France harboring the V367F mutation, which was previously reported to enhance affinity to the ACE2 receptor.
On comparison, selection pressure analysis of the SARS-CoV-2 S protein revealed a single positively selected residue located in the S1 domain; the head region of the S protein, suggesting the role of diversifying selection during the current stage of the pandemic.
The presence of 29 negatively selected sites in only the European group of strains may also have a role in the viral fitness in that group. To comment on whether the evidence of purifying selection observed only in the European strains could influence viral fitness sufficient to be correlated with increased disease severity in Europe would be premature as such, a larger number of sequences is needed to substantiate this claim. Nevertheless, we identified several (n=29) negatively selected sites in the S protein, limited only to the European strains. Mutational analysis of the study strains revealed a D614G mutation in approximately 60% of the sequences. Geographical mapping of these variants revealed a maximum proportion of variants in Europe (63/101). The D614G S protein variant has been correlated with fatal outcomes in the European population in the first quarter of the year. Analysis of 86 SARS-CoV-2 genomes9 revealed 14 nucleotide and 8 missense mutations (F32I), (H49Y), (S247R), (N354D), (D364Y), (V367F), (D614G), (P1143L) located in the RBD region of the S glycoprotein. Such mutations may induce conformational changes (open status) and expose the cleavage domain of the spike protein to FURIN, or TMPRSS2 thereby enhancing its cleavage. Variability of SARS-CoV-2 mutations, may contribute to the emergence of different phylogenetic clades which may explain the disparity in death rate. However, both the host and viral genetic factors, along with the geographical distribution have been associated with the high and low COVID-19 related fatalities.10
Findings of our study are supported by those from Eaaswarkhanth et al.,11, who reported the D614G variant in European populations from Spain, Italy, France, Switzerland, Belgium, and the Netherlands resulting in a high mortality rate. However, this evidence is circumstantial and may vary across different geographical populations. Among different European countries, high case fatality rate and G614 mutated viral strains have been strongly correlated. During the first quarter of this year, the D614G S protein variant was correlated with fatal outcomes in European populations and countries including Italy, France, Belgium, and Spain. Surprisingly, investigators identified 12 positive selective pressure sites in 2002 SARS-CoV outbreak in China, including 613 in domain 1 of the S protein, which is a shift of one codon from SARS-CoV-2. We observed D614G variants under high selective pressure in Asian and European strains; however, the difference was not statistically significant in the US strains. In addition, the presence of 29 negatively selected codon sites under low selection pressure in the European group may imply viral fitness compared with circulating strains in other continents. Thus, it appears that selective pressure-driven fitness with high substitution rates in the S protein may facilitate SARS-CoV-2 adaptive evolution and its widespread transmission as well as its persistence in the community.
Since the emergence of SARS-CoV-2 in late December 2019, the epidemiology and associated mortality have varied depending on the countries affected. During the first quarter of this year, the D614G S spike protein variant was correlated with fatal outcomes in European populations, including countries such as Italy, France, Belgium, and Spain. We report D614G variants under high selective pressure in Asian and European strains. In addition, the presence of 29 negatively selected codon sites under low selection pressure in the European group may imply improved viral fitness with strains circulating in other continents. Therefore, selective pressure influences the S protein with the maximum substitution rate, which may facilitate adaptive evolution of the virus, and contribute to the worldwide spread of the virus.
ACKNOWLEDGMENTS
The authors acknowledge the support from the Department of Virology, Postgraduate Institute of Medical Education and Research, India.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
AUTHORS’ CONTRIBUTION
SS conceptualized the research, designed the experiment, analyzed the data and wrote the manuscript. VT analyzed the data, prepared and edited the manuscript. RKR conceptualized the research and edited the manuscript. SKS edited the manuscript. All the authors approved the final version of the manuscript.
FUNDING
None.
ETHICS STATEMENT
Not applicable.
AVAILABILITY OF DATA
The datasets generated and analysed during the current study are available from the corresponding author on reasonable request.
- WHO Coronavirus Disease (COVID-19). 2021. https://covid19.who.int/
- Su YCF, Bahl J, Joseph U, et al. Phylodynamics of H1N1/2009 influenza reveals the transition from host adaptation to immune-driven selection. Nat Commun. 2015;6(1):7952.
Crossref - Zhou P, Yang X-L, Wang X-G, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270-273.
Crossref - Tang X, Wu C, Li X, et al. On the origin and continuing evolution of SARS-CoV-2. Natl Sci Rev. 2020;7(6):1012-1023.
Crossref - Zhang CY, Wei JF, He SN. Adaptive evolution of the spike gene of SARS coronavirus: Changes in positively selected sites in different epidemic groups. BMC Microbiol. 2006;6:88.
Crossref - Hoffmann M, Kleine-Weber H, Schroeder S, et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020;181(2):271-280.
Crossref - Laamarti M, Alouane T, Kartti S, et al. Large scale genomic analysis of 3067 SARS-CoV-2 genomes reveals a clonal geo-distribution and a rich genetic variations of hotspots mutations. PloS One . 2020;15(11):e240345.
Crossref - Ou J, Zhou Z, Dai R, et al. Emergence of RBD mutations in circulating SARS-CoV-2 strains enhancing the structural stability and human ACE2 receptor affinity of the spike protein. BioRxiv. 2020.
- Phan T. Genetic diversity and evolution of SARS-CoV-2. Infect Genet Evol. 2020;81:104260.
Crossref - Onder G, Rezza G, Brusaferro S. Case-Fatality Rate and Characteristics of Patients Dying in Relation to COVID-19 in Italy. JAMA – Journal of the American Medical Association. American Medical Association. 2020;323:1775-1776.
Crossref - Eaaswarkhanth M, Al Madhoun A, Al-Mulla F. Could the D614G substitution in the SARS-CoV-2 spike (S) protein be associated with higher COVID-19 mortality? Int J Infect Dis. 2020;96:459-460.
Crossref - Becerra-Flores M, Cardozo T. SARS-CoV-2 viral spike G614 mutation exhibits higher case fatality rate. Int J Clin Pract. 2020;74(8):e13525.
Crossref
© The Author(s) 2022. Open Access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License which permits unrestricted use, sharing, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.