


ISSN: 0973-7510
E-ISSN: 2581-690X
Helicobacter pylori is an important causative agent of gastrointestinal and hepatobiliary diseases. The aim of this study was to investigate phylogenetic relationships among H. pylori strains from the oral cavities of healthy individuals, gastric biopsies from gastroduodenal (GI) patients and bile samples from hepatobiliary (HB) patients in the northeast of Thailand. The DNA sequences of a portion of the vacuolating cytotoxin A gene (vacA) were investigated. Phylogenetic trees were constructed using the neighbor-joining and maximum-likelihood methods. The vacA sequences of H. pylori fell into four main groups on the trees. Strains from healthy persons fell into two widely separated but well-supported groups, while most H. pylori vacA sequences from HB patients were distributed in another two groups. In contrast, the H. pylori strains from GI patients were scattered across the tree, without a clear geographical pattern. In conclusion, the sequence of vacA may be useful to classify the genetic relationship of H. pylori derived from different sources.
H. pylori, vacuolating cytotoxin A gene, Gastrointestinal diseases, hepatobiliary diseases and healthy persons.
Helicobacter pylori is a Gram-negative spiral-shaped bacterium, a causative agent of many gastrointestinal diseases such as dyspepsia, gastritis, peptic ulcer and gastro-esophageal reflux disease, which can lead to gastric adenocarcinoma.1 This pathogen has also been recognized as a type I carcinogen by the World Health Organization’s International Agency for Research on Cancer because it is associated with gastric mucosa-associated lymphoid tissue lymphoma and gastric adenocarcinoma2.
Helicobacter pylori was recently proposed to be associated with diseases outside the gastroduodenal tract, particularly the hepatobiliary duct system.3 Such diseases include cholangiocarcinoma (CCA), which is a primary cancer of the biliary epithelium and is highly endemic in Northeast Thailand.4 Several studies have demonstrated that the virulence genes in H. pylori, including the vacuolating cytotoxin gene A (vacA) and cytotoxin-associated gene A (cagA), may help to predict the association between H. pylori infection and clinical outcomes4,5. However, cagA and other virulence genes (viz. babA, cagE and iceA) are present in only some strains of H. pylori, whereas vacA is present in all H. pylori strains.6
Vacuolating cytotoxin induces intracellular vacuoles, leading to epithelial damage of eukaryotic cells7 by suppressing epithelial proliferation, promoting apoptotic cell death,8 inducing cytoskeletal changes8 and suppressing epithelial proliferation.9 Each allelic variant of vacA contains one of two classes of signal-region variant (s1 or s2) and one of two classes of middle region (m1 or m2) variant.10 Allelic variation among strains results in variation of vacuolating activity.11 The s1/m1 H. pylori strain displays a higher vacuolating activity than that does the s1/m2 strain and might be associated with gastritis and gastric adenocarcinoma. In contrast, the vacA s2/m2 strain is rarely associated with such diseases due to the absence of cytotoxic activity.12 Although, vacA genotype status might help predict the development of diseases, one report has suggested that genotype of vacA was not associated with the clinical outcome of peptic ulcer, duodenal ulcer and gastric cancer.13 Therefore, in this study, we test the idea that the vacA sequence can be used to distinguish among clinical sources of specimens.
Routes of H. pylori transmission are oral-oral,14 gastro-oral15 and fecal-oral.16 Saliva can be a reservoir of H. pylori, potentially infecting or reinfecting the stomach.17 It might be that the genotypes of H. pylori strains in saliva of healthy persons can be useful for mapping the geographical distribution of H. pylori strains associated with gastroduodenal (GI) and hepatobiliary (HB) diseases. To explore this possibility, we sequenced portions of the vacA gene from specimens from healthy individuals and from patients with various gastroduodenal and hepatobiliary diseases.
Study subjects
Study subjects consisted of healthy persons, patients with GI diseases and patients with hepatobiliary diseases admitted to Srinagarind Hospital, Faculty of Medicine, Khon Kaen University, Thailand. Approval for the study was obtained from the Human Ethics Committee of Khon Kaen University (approval nos. HE571489 and HE581271) and prior written informed consent was obtained from all participants.
Collection and processing of saliva samples
Thirty saliva samples were collected from healthy person using the method of Silva et. al. (2009), with slight modifications.18 Two ml of each saliva sample was added to 8ml of Brucella broth (CRITERION™, USA) and incubated for 3 days at 37°C with shaking under microaerobic atmosphere (5% O2, 10% CO2 and 85% N2). After centrifugation at 13,000 rpm for 10 minutes at 4°C, the pellet from each culture was used as a source of DNA for amplification of vacA by nested PCR.
Collection and processing of gastric biopsy and bile specimens
Gastric biopsies were performed on dyspeptic patients undergoing gastro-endoscopic examinations at the Endoscopy Unit, Srinagarind Hospital, Faculty of Medicine, Khon Kaen University. Dyspeptic patients (GI) were characterized as having non-ulcer disease (NUD) (n = 20), peptic ulcer disease (PUD) (n = 3) or gastric cancer (GC) (n = 2). Gastric samples from these patients were positive for the rapid urease test indicating H. pylori infection. Bile samples, positive for H. pylori via PCR detection of ureA gene, were taken from patients with hepatobiliary disorders, namely, CCA (n = 20) and cholelithiasis (n = 9), at the Surgical Unit, Srinagarind Hospital, Faculty of Medicine, Khon Kaen University. The sources of the clinical specimens are summarized in Table 1.
Table (1):
Sources of samples used in this study.
| Source of samples | Number of samples | Sample code |
|---|---|---|
| Healthy persons (n=30) • Saliva samples |
30 | – |
| Gastroduodenal patients (n=25) • Non-ulcer disease (NUD) |
20 | – |
| • Peptic ulcer diseases (PUD) | 3 | – |
| • Gastric cancer (GC) | 2 | – |
| Hepatobiliary patients (n=30) • Cholangiocarcinoma (CCA) |
20 | – |
| • Cholelithiasis | 9 | – |
| • Other hepatobiliary diseases | 1 | – |
| Reference strains* | ||
| Japan | 5 | CP06826, AB190972, AB190973, AF071097, AB190965 |
| India | 3 | GQ331975, GQ331980, GQ331984 |
| Kenya | 3 | AF191641, AF191642, AF191644 |
| USA | 2 | CP003474, AE000511 |
| China | 1 | CP003419 |
| Italy | 1 | U95971 |
| Western Africa | 1 | CP002571 |
* = All reference strains taken from Genbank were isolated from GI patients
DNA extraction
DNA was extracted from pellets of saliva samples using a modified method based on the Puregene DNA Purification System (Gentra System, USA).19 DNA was extracted from bile samples and biopsy specimens using the Gentra System DNA extraction and purification kit (Big Lake, MN, USA), respectively, as previously described4,20.
PCR amplification and DNA Sequencing
Amplification of a portion of the vacA gene from each sample was performed by nested PCR using primers and conditions as shown in Table 2. The inner primers (forward: nt. 2673-2692, reverse: nt. 2929-2948; GenBank GQ331975) amplified a fragment of vacA encoding amino acid positions 892 to 979. The region amplified does not include the S or the M region. Nested PCR assay was performed in a total volume of 25µl containing 500ng of DNA template, 0.2 mM dNTPs (Amresco, Ohio, USA), 1X PCR buffer (10 mM Tris-HCl pH 8.3, 50mM KCl, 0.1 mg/ml bovine serum albumin, 10mM (NH4)2SO4, and 1.5 mM MgCl2) (RBC bioscience, Taipei, Taiwan), 0.5 U Taq polymerase (RBC Bioscience, Taipei, Taiwan), and 0.4 µM vacA primers. Thermocycling was conducted in a C1000™ Thermal Cycler (BioRad, USA). Amplicons were purified using spin column PCR clean up kit (DNA sequencing service; BIONEER, Korea) and sequenced using Sanger sequencing method (DNA sequencing service; BIONEER, Korea).
Table 2:
Primer sequences and PCR condition for detection of H. pylori.
| Specific for | Primer sequence (5’>>>3’) | Product size (bp) | PCR condition |
|---|---|---|---|
| vacA19 (H. pylori) |
OF-GCATGATTTTGGCACCATTG | 429 | 95°C 30 s, 54°C 30 s, 72°C 45 s (35 cycles) |
| OR-TTTTCATATTTAGGGGCAAA | |||
| IF-GCATGATTTTGGCACCATTG | 276 | 95°C 30 s, 62°C 30 s, 72°C 45 s (35 cycles) |
|
| IR-ATCGCATTGCTCAAGCTCAA | |||
| vacA s1/s224 | F-ATGGAAATACAACAAACACAC | 259/286 | 94°C 60 s, 58°C 60 s, 72°C 60 s (35 cycles) |
| R-CTGCTTGAATGCGCCAAAC | |||
| vacA m1/m224 | F-CAATCTGTCCAATCAAGCGAG | 567/642 | 94°C 60 s, 55°C 60 s, 72°C 60 s (35 cycles) |
| R-GCGTCAAAATAATTCCAAGG |
Phylogenetic analysis
The vacA sequences from all samples, including sixteen reference sequences of H. pylori vacA from GenBank (Table 1), were aligned using the MAFFT program (http://www.ebi.ac.uk/Tools/msa/mafft). The aligned sequences were adjusted and edited using BioEdit v.7.0.5.2. Phylogenetic analyses were conducted in MEGA software version 7.021. The phylogenetic trees were constructed and compared using the neighbor-joining and maximum-likelihood methods with 1,000 bootstrap replicates. The Kimura 2-parameter model with Gamma distribution was used as the best model (the model giving the best log-likelihood value.
The phylogenetic trees (Fig. 1 and 2) revealed that most sequences from Thai samples fell into distinct clades and were generally also distinct from sequences from other countries. The alignment of 101 partial sequences of the vacA gene was 276 bp in length, and translated to 88 amino acids (Fig. 3).
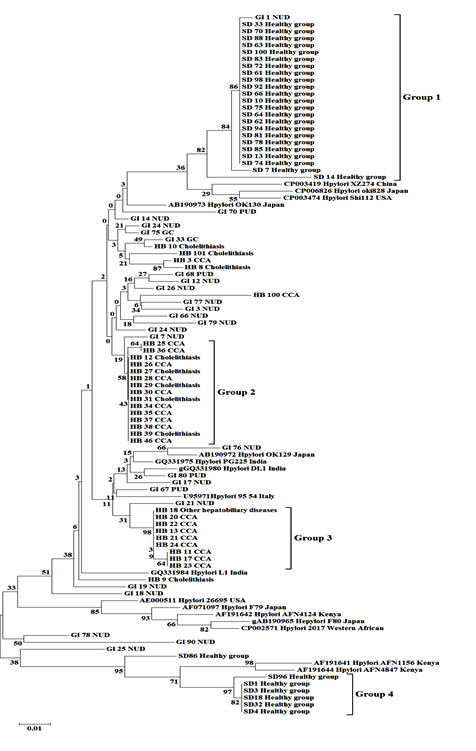 Fig. 1.Neighbor joining tree with 1,000 bootstraps from a dataset of partial vacA gene sequences. Numbers on the branches are bootstrap values (%). Disease conditions are represented by NUD; nonpeptic ulcer diseases, PUD; peptic ulcer diseases and CCA; cholangiocarcinoma
Fig. 1.Neighbor joining tree with 1,000 bootstraps from a dataset of partial vacA gene sequences. Numbers on the branches are bootstrap values (%). Disease conditions are represented by NUD; nonpeptic ulcer diseases, PUD; peptic ulcer diseases and CCA; cholangiocarcinoma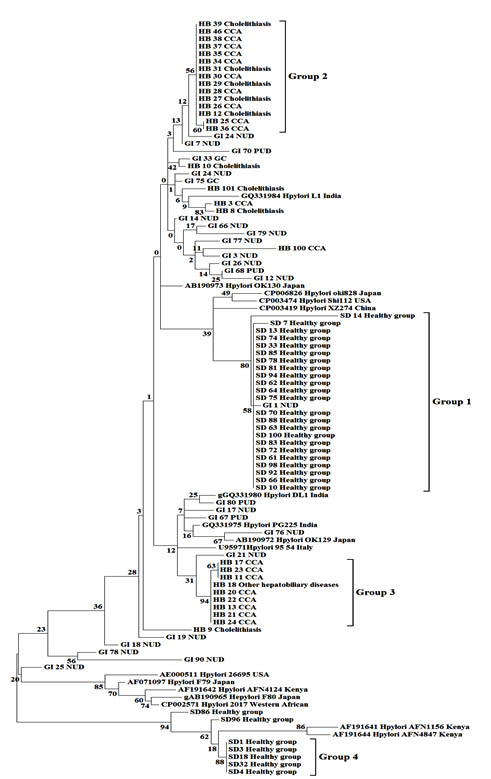
Fig. 2.Maximum likelihood tree with 1,000 bootstraps from a dataset of partial vacA gene sequences. Numbers on the branches are bootstrap values (%). Disease conditions are represented by NUD; nonpeptic ulcer diseases, PUD; peptic ulcer diseases and CCA; cholangiocarcinoma
The neighbor-joining tree (Fig. 1) placed the thirty sequences from healthy Thai people into two very distinct groups with high bootstrap support: group 1 (82% support) consisted of 23 sequences and group 4 (97% support) included 6 sequences. Sequences from Thai hepatobiliary patients mostly fell into two groups, group 2 consisting of 10 sequences from CCA patients together with 5 sequences from Thai gall-stone patients (58% bootstrap support), and group 3 consisting of 8 sequences from CCA patients and 1 sequence from patients with other hepatobiliary diseases (98% support). In contrast, most vacA sequences from Thai GI patients were not close to those from healthy individuals or HB patients, but were scattered across the tree and often among vacA sequences of GI patients from various countries. Similarly, H. pylori sequences from saliva samples of healthy persons, gastric biopsies of gastrointestinal tract patients (GI) and bile samples of hepatobiliary patients were grouped in the maximum likelihood tree (Fig. 2) in a manner consistent with the neighbor joining tree. Bootstrap values differed slightly between the trees and the 4 groups, while still strongly supported, fell in somewhat different parts of the two trees.
In this study, we also determined the alleles of vacA (s and m) present in the bile samples of hepatobiliary patients which are nearly all of were s1 and m1 (data not shown). Therefore, these alleles can’t be used to identify the different clinical origins of samples.
Translated amino acid sequences were compared among the four groups of H. pylori (indicated in Fig. 1 and 2) and found to differ at many positions (Fig. 3).
 Fig. 3. Differences among the source-groups of H. pylori strains in an 88-amino acid portion translated from the vacA gene sequence. H. pylori PG225 is the reference strain from the NCBI database (GQ331975). Abbreviation of amino acids represented by A, Alanine; C, Cysteine; D, Aspartic acid; E, Glutamic acid; G, Glycine; H, Histidine; I, Isoleucine; K, Lysine; L, Leucine; N, Asparagine; Q, Glutamine; R, Arginine; S, Serine; T, Threonine; V, Valine
Fig. 3. Differences among the source-groups of H. pylori strains in an 88-amino acid portion translated from the vacA gene sequence. H. pylori PG225 is the reference strain from the NCBI database (GQ331975). Abbreviation of amino acids represented by A, Alanine; C, Cysteine; D, Aspartic acid; E, Glutamic acid; G, Glycine; H, Histidine; I, Isoleucine; K, Lysine; L, Leucine; N, Asparagine; Q, Glutamine; R, Arginine; S, Serine; T, Threonine; V, ValineOur study is the first investigation on the genetic diversity and relationships of vacA gene sequences of H. pylori from different sources; saliva from healthy persons, gastric biopsies from gastrointestinal tract patients (GI) and bile samples from hepatobiliary patients (CCA and cholelithiasis) in Thailand. Partial vacA gene sequences from healthy persons were obviously very different from those of HB patients. H. pylori strains from saliva samples of healthy persons fell into two well-supported but widely separated clades (clades 1 and 4). Strains isolated from HB patients, especially those with CCA, mostly also fell into distinct clades (clades 2 and 3). These results indicate that particular genotypes of vacA may occur in strains of H. pylori specific to particular sources (disease status of the host and geography). This applied strongly to strains from CCA patients: this disease is highly endemic in Thailand. Interestingly, most H. pylori strains from GI tracts of Thai patients were distributed among other strains of GI patients from different countries, but not among the strains from healthy persons or HB patients. It seems that most H. pylori strains in saliva of healthy persons in Northeast Thailand may not be derived from the strains isolated from GI and HB patients. The partial vacA gene sequence developed in this study has the potential to distinguish among H. pylori strains from different origins and sources. In this study, genetic diversity and relationships of H. pylori from different sources were determined using neighbor-joining and maximum-likelihood methods. Results from the two methods were in broad agreement, suggesting we were close to the true phylogenetic tree. An advantage of the neighbor-joining method is more computational efficiency and speed than maximum-likelihood method, making it suitable for analysis of large data sets. However, this method is unsuitable if very divergent sequences are included.22 The maximum-likelihood (ML) approach is character-based and is now widely used owing to the development of increasingly realistic and explicit models of sequence evolution. However, likelihood calculations and tree searches under the likelihood criterion are computationally demanding, making this approach very slow, especially if many sequences are included in the alignment.22 Researchers can choose the method that best suits their data.24
Differences were apparent between the four clades of sequences at both the DNA and the amino acid level. Presumably, this is a consequence of adaptation to different environmental sources, hosts and geography.13,23 However, the relationship between vacA diversity, origin, and disease state in the host should be further investigated.
In conclusion, these findings support that the partial vacA sequence may be useful as a marker to distinguish H. pylori from difference sources, such as from healthy persons or patients with gastrointestinal and hepatobiliary diseases, and as a marker to investigate genetic relationships among the strains isolated from different sources.
ACKNOWLEDGMENTS
This study was supported by the Higher Education Research Promotion and National Research University Project of Thailand, Office of the Higher Education Commission, through the Center of Excellence in Specific Health Problems in Greater Mekong Sub-region cluster (SHeP-GMS), Khon Kaen University to Chariya Chomvarin et al. The authors thank the Liver Fluke and Cholangiocarcinoma Research Center for a scholarship. We would like to acknowledge Prof. David Blair, Publication Clinic, Khon Kaen University for editing the manuscript via Publication Clinic, Khon Kaen University, Thailand.
- Peek, R.M., Jr., Blaser, M.J. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nature reviews Cancer, 2002; 2: 28-37.
- Moss, S.F., Malfertheiner, P. Helicobacter and gastric malignancies. Helicobacter, 2007; 12 Suppl 1: 23-30.
- Rabelo-Goncalves, E.M., Roesler, B.M., Zeitune, J.M. Extragastric manifestations of Helicobacter pylori infection: Possible role of bacterium in liver and pancreas diseases. World J Hepatol, 2015; 7: 2968-79.
- Boonyanugomol, W., Chomvarin, C., Sripa, B., Bhudhisawasdi, V., Khuntikeo, N., Hahnvajanawong, C., Chamsuwan, A. Helicobacter pylori in Thai patients with cholangiocarcinoma and its association with biliary inflammation and proliferation. HPB (Oxford), 2012; 14: 177-84.
- Boonyanugomol, W., Chomvarin, C., Sripa, B., Chau-In, S., Pugkhem, A., Namwat, W., Wongboot, W., Khampoosa, B. Molecular analysis of Helicobacter pylori virulent-associated genes in hepatobiliary patients. HPB (Oxford), 2012; 14: 754-63.
- Dunn, B.E., Cohen, H., Blaser, M.J. Helicobacter pylori. Clin Microbiol Rev, 1997; 10: 720-41.
- Reyrat, J.M., Rappuoli, R., Telford, J.L. A structural overview of the Helicobacter cytotoxin. Int J Med Microbiol, 2000; 290: 375-9.
- Blaser, M.J., Atherton, J.C. Helicobacter pylori persistence: biology and disease. J Clin Invest, 2004; 113: 321-33.
- Pai, R., Cover, T.L., Tarnawski, A.S. Helicobacter pylori vacuolating cytotoxin (VacA) disorganizes the cytoskeletal architecture of gastric epithelial cells. Biochem Biophys Res Commun, 1999; 262: 245-50.
- Miernyk, K., Morris, J., Bruden, D., McMahon, B., Hurlburt, D., Sacco, F., Parkinson, A., Hennessy, T., Bruce, M. Characterization of Helicobacter pylori cagA and vacA genotypes among Alaskans and their correlation with clinical disease. J Clin Microbiol, 2011; 49: 3114-21.
- Atherton, J.C., Cao, P., Peek, R.M., Jr., Tummuru, M.K., Blaser, M.J., Cover, T.L. Mosaicism in vacuolating cytotoxin alleles of Helicobacter pylori. Association of specific vacA types with cytotoxin production and peptic ulceration. J Biol Chem, 1995; 270: 17771-7.
- Atherton, J.C. The pathogenesis of Helicobacter pylori-induced gastro-duodenal diseases. Annu Rev Pathol, 2006; 1: 63-96.
- Miftahussurur, M., Sharma, R.P., Shrestha, P.K., Suzuki, R., Uchida, T., Yamaoka, Y. Molecular Epidemiology of Helicobacter pylori Infection in Nepal: Specific Ancestor Root. PLoS One, 2015; 10: e0134216.
- Al Sayed, A., Anand, P.S., Kamath, K.P., Patil, S., Preethanath, R.S., Anil, S. Oral Cavity as an Extragastric Reservoir of Helicobacter pylori. ISRN Gastroenterol, 2014; 2014.
- Luzza, F., Mancuso, M., Imeneo, M., Contaldo, A., Giancotti, L., Pensabene, L., Doldo, P., Liberto, M.C., Strisciuglio, P., Foca, A., Guandalini, S., Pallone, F. Evidence favouring the gastro-oral route in the transmission of Helicobacter pylori infection in children. Eur J Gastroenterol Hepatol, 2000; 12: 623-7.
- Mitipat, N., Siripermpool, P., Jadwattanakul, T., Chaunthongkum, S. The prevalence of Helicobacter pylori infection in patients with gastrointestinal symptoms in Chon Buri, Thailand. Southeast Asian J Trop Med Public Health, 2005; 36: 341-6.
- Burgers, R., Schneider-Brachert, W., Reischl, U., Behr, A., Hiller, K.A., Lehn, N., Schmalz, G., Ruhl, S. Helicobacter pylori in human oral cavity and stomach. Eur J Oral Sci, 2008; 116: 297-304.
- Silva, D.G., Stevens, R.H., Macedo, J.M., Albano, R.M., Falabella, M.E., Veerman, E.C., Tinoco, E.M. Detection of cytotoxin genotypes of Helicobacter pylori in stomach, saliva and dental plaque. Arch Oral Biol, 2009; 54: 684-8.
- Tirapattanun, A., Namwat, W., Kanthawong, S., Wongboot, W., Wongwajana, S., Wongphutorn, P., Chomvarin, C. Detection of Heicobacter pylori and virulence-associated genes in saliva samples of asymptomatic person in Northeast of Thailand. Southeast Asian J Trop Med Public Health 2016; 47: 1246-56.
- Chomvarin, C., Namwat, W., Chaicumpar, K., Mairiang, P., Sangchan, A., Sripa, B., Tor-Udom, S., Vilaichone, R.K. Prevalence of Helicobacter pylori vacA, cagA, cagE, iceA and babA2 genotypes in Thai dyspeptic patients. Int J Infect Dis, 2008; 12: 30-6.
- Kumar, S., Stecher, G., Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol Biol Evol, 2016; 33: 1870-4.
- Bruno, W.J., Socci, N.D., Halpern, A.L. Weighted neighbor joining: a likelihood-based approach to distance-based phylogeny reconstruction. Mol Biol Evol, 2000; 17: 189-97.
- Kodaman, N., Pazos, A., Schneider, B.G., Piazuelo, M.B., Mera, R., Sobota, R.S., Sicinschi, L.A., Shaffer, C.L., Romero-Gallo, J., de Sablet, T., Harder, R.H., Bravo, L.E., Peek, R.M., Jr., Wilson, K.T., Cover, T.L., Williams, S.M., Correa, P. Human and Helicobacter pylori coevolution shapes the risk of gastric disease. Proc Natl Acad Sci U S A, 2014; 111: 1455-60.
- Qiao, W., Hu, J.L., Xiao, B., Wu, K.C., Peng, D.R., Atherton, J.C., Xue, H. cagA and vacA genotype of Helicobacter pylori associated with gastric diseases in Xi’an area. World J Gastroenterol, 2003; 9: 1762-6.
© The Author(s) 2017. Open Access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License which permits unrestricted use, sharing, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.