


ISSN: 0973-7510
E-ISSN: 2581-690X
The present research explores the urease inhibitory potential of a designed set of quinoline-2-one/chalcone hybrid derivatives (4a-e). The biological assessment demonstrated that these molecules exhibit strong urease inhibition, with IC50 values ranging from 0.58-1.90 µM. Their inhibitory potency notably surpasses that of the reference compound, thiourea (IC50 = 21.50 µM). Within this series, compounds 4a, 4c, and 4e emerged as the most active inhibitors, displaying IC50 values of 0.93 ± 0.08 µM, 0.75 ± 0.06 µM, and 0.58 ± 0.04 µM, respectively. In addition, the anti-Helicobacter pylori potential of these hybrids was assessed against three metronidazole-resistant H. pylori strains using the disk diffusion technique. The outcomes revealed a strong association, indicating that methoxy-bearing analogs 4c and 4e identified as the most effective urease inhibitors also exhibited pronounced antibacterial action against H. pylori. Collectively, these observations underscore the promising dual therapeutic profile of this hybrid framework.
Urease, Helicobacter pylori, Antibacterial, Paper Disk, Inhibition Zone
Urease is a nickel-dependent metalloenzyme responsible for catalyzing the hydrolysis of urea into ammonia and carbamate.1,2 This enzyme is structurally diverse and occurs in multiple isoforms identified across a wide range of biological systems, including plants, fungi, and numerous bacterial species.3,4 Within pathogenic microorganisms, urease functions as a major virulence determinant, critically contributing to the onset and progression of various disorders such as peptic ulcer disease, gastric carcinoma, urinary tract infections, hepatic encephalopathy, and several renal complications.5
A notable example is Helicobacter pylori, a microaerophilic bacterium characterized by exceptionally high urease activity. This pathogen is one of the most common bacterial inhabitants of the human stomach, colonizing more than half of the global population.6 The urease enzyme produced by H. pylori enables the organism to survive within the stomach’s acidic environment by generating ammonia, which forms a protective alkaline microenvironment surrounding the bacterium.7 The resulting accumulation of ammonia damages the gastroduodenal epithelium, contributing to the development of severe gastrointestinal conditions such as duodenal ulcers, gastric adenocarcinomas, and lymphomas, affecting approximately 15%-20% of infected patients.8,9 Supporting this, Eaton et al. demonstrated that urease-deficient mutant strains of H. pylori failed to colonize the stomachs of gnotobiotic piglets,10,11 confirming the enzyme’s indispensable role in bacterial survival and pathogenesis. Consequently, the design and discovery of potent urease inhibitors are of substantial therapeutic relevance.
In recent years, significant attention has been directed toward the antimicrobial potential of quinoline-containing scaffolds, as many derivatives have shown enhanced biological performance compared with conventional agents.12-15 For instance, Elshaier et al. reported the synthesis and biological evaluation of a novel group of quinoline derivatives as urease inhibitors, identifying Compound I (Figure 1) as the most active molecule in the series with an IC50 of 1.83 µM.16 Similarly, Elbastawesy et al. developed a distinct quinoline-based collection of compounds, among which Compound II (Figure 1) exhibited strong urease inhibition, with an IC50 value of 0.46 µM.17
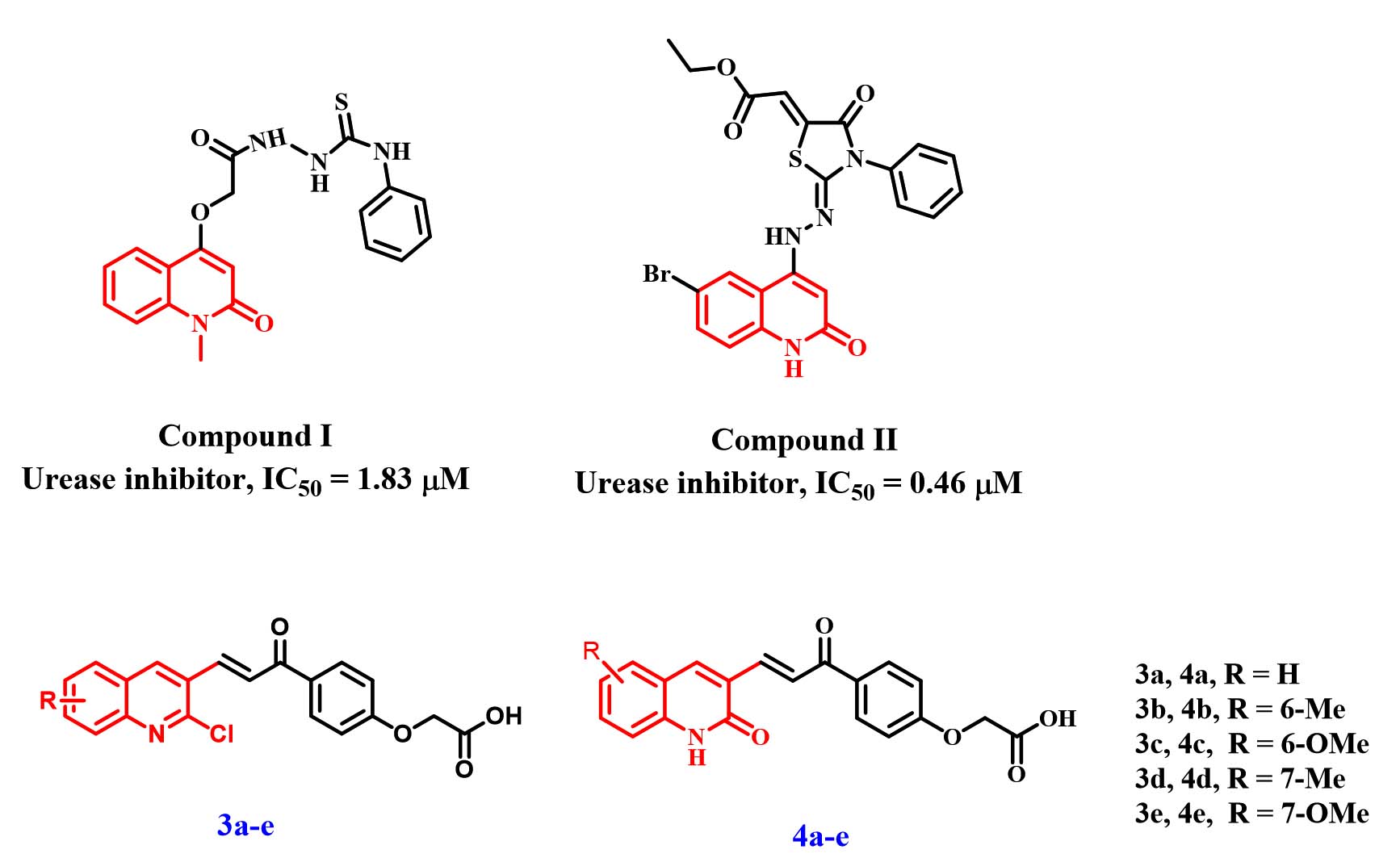
Figure 1. Structures of quinoline-based urease inhibitors I, II and target compounds 3a-e and 4a-e
Chalcones, chemically defined as 1,3-diaryl-2-propen-1-ones, represent another prominent class of naturally occurring molecules widely recognized for their diverse pharmacological activities, notably their antimicrobial potential.18-20 Their natural abundance and the relative simplicity of their synthesis have generated significant interest in utilizing chalcone frameworks for the creation of new therapeutic leads.21-23
At the same time, molecular hybridization, a strategy that combines two or more pharmacologically active moieties within a single molecular structure, has emerged as a powerful tool for developing multifunctional bioactive compounds. Such hybrid architectures can integrate distinct mechanisms of action, often resulting in synergistic or dual biological properties that enhance therapeutic efficacy against pathogenic microorganisms.21,24-27
In our previous work28 we described the design and synthesis of a novel library of quinoline–chalcone hybrids (3a-e and 4a-e, Figure 1), created by uniting these two privileged structural motifs into a single molecular entity. Upon biological screening, compounds within the 3a-e subset demonstrated the most pronounced antibacterial activities against a panel of six bacterial strains.
Encouraged by the promising antibacterial outcomes obtained from the 4a-e series (Figure 1) and inspired by the well-documented urease-inhibitory potential of quinoline derivatives such as Compounds I and II (Figure 1), the present study was designed to explore the urease inhibitory capabilities of compounds 4a-e. Moreover, this work further investigates the anti-Helicobacter pylori efficacy of these hybrid molecules to assess their broader therapeutic potential.
Experimental
Urease inhibition assay
The in vitro urease inhibitory potential of the synthesized compounds was examined using Jack bean urease, according to the Berthelot phenol assay protocol.29,30 This colorimetric method quantifies the amount of ammonia released during the enzymatic hydrolysis of urea, thereby reflecting the enzyme’s catalytic activity. All assays were performed in triplicate, and results are reported as the mean ± standard deviation (SD). A comprehensive and sequential description of the experimental procedure is provided in the Supplementary file.
Bacterial growth inhibition assay (disk diffusion method)
The antibacterial activity of the prepared compounds against Helicobacter pylori was determined using the standard filter paper disk diffusion approach. Experimental assays were performed on Brucella agar medium supplemented with 7% defibrinated horse blood and incubated at 37 °C under microaerophilic conditions. Dimethyl sulfoxide (DMSO) was utilized as the dissolution medium for all tested samples.31 Each disk diffusion test was conducted in triplicate. The diameters of the inhibition zones were analyzed using One-way ANOVA to determine significant differences between the synthesized compounds and the positive control, with a P-value <0.05 defined as the threshold for significance. The complete and detailed experimental protocol is outlined in the Supplementary file.
Chemistry
The intended quinoline–chalcone hybrid derivatives (3a-e and 4a-e) were efficiently prepared following established synthetic routes previously reported in the literature,28 as illustrated in Figure 2. Structural elucidation and confirmation of product identity were accomplished by comparing their physical characteristics and spectroscopic profiles (¹H NMR and ¹³C NMR) with the corresponding literature data, ensuring complete consistency with reported values.
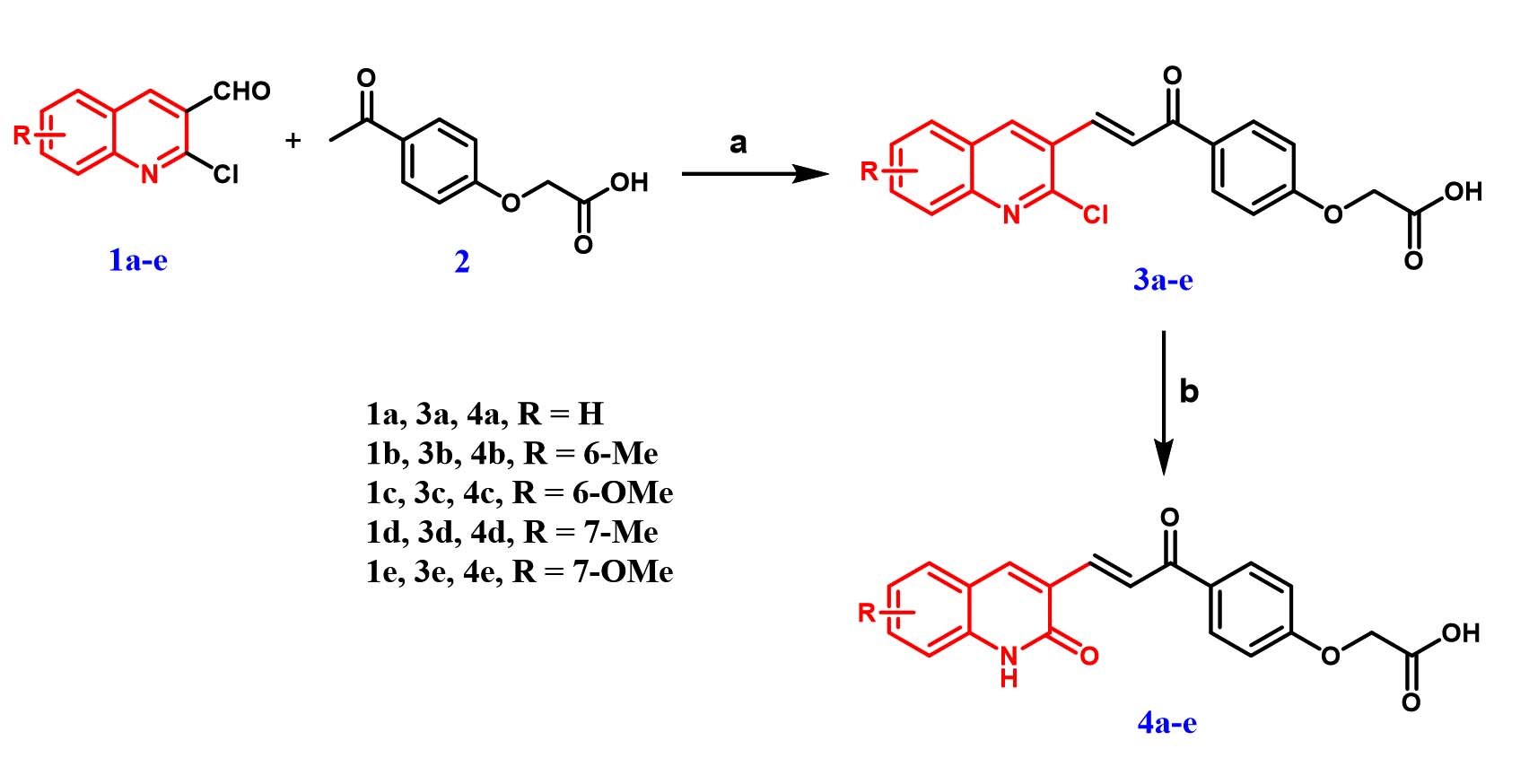
Figure 2. Synthesis of target compounds 3a-e and 4a-e
Reagent and Reactions conditions: (a) Ethanol, 10% NaOH, r.t. 15 mins, 79%-82% (b) Acetic acid, reflux, 6 hrs, 68%-75%.
Biology
Urease inhibition assay
The newly synthesized quinoline-based hybrid molecules (4a-e) were assessed for their inhibitory efficacy against the urease enzyme in vitro, using thiourea as the reference standard inhibitor.29 The obtained results, represented as IC50 values, are summarized in Table 1.
Table (1): IC50 of compounds 4a-e against urease
 |
||
|---|---|---|
| Compd. No. | R | Urease inhibition (IC50, µM) |
| 4a | H | 0.93 ± 0.08 |
| 4b | 6-Me | 1.90 ± 0.15 |
| 4c | 6-OMe | 0.75 ± 0.06 |
| 4d | 7-Me | 1.45 ± 0.10 |
| 4e | 7-OMe | 0.58 ± 0.05 |
| Thiourea | – | 21.50 ± 0.80 |
All tested compounds exhibited notable urease inhibition, with IC50 values ranging between 0.58 µM and 1.90 µM, indicating substantial potency. This inhibitory activity was significantly superior to that of the benchmark compound thiourea (IC50 = 21.50 µM). Among the series, compound 4e (R = 7-OMe) emerged as the most powerful inhibitor, displaying an IC50 of 0.58 ± 0.05 µM approximately 37 times more active than thiourea. The inhibitory efficiency was strongly affected by the position of the methoxy substituent: compound 4c (R = 6-OMe, IC50 = 0.75 µM) was about 1.3-fold less active than 4e. The unsubstituted derivative 4a (R = H, IC50 = 0.93 ± 0.08 µM) showed slightly lower potency, being 1.6 and 1.3 times less effective than 4e and 4c, respectively.
A marked decline in inhibitory capability was detected when the methoxy functionality was replaced by a methyl group. This structural modification generated compounds 4d (R = 7-Me, IC50 = 1.45 ± 0.10 µM) and 4b (R = 6-Me, IC50 = 1.90 ± 0.15 µM), which were the least potent members of the series. Specifically, 4d displayed 2.5-fold lower activity compared with its methoxy analogue (4e), whereas 4b was 2.6-fold weaker than 4c. Collectively, these findings demonstrate that both the electronic effects and the positional orientation of substituents on the quinoline phenyl ring play a decisive role in determining urease inhibition. The established structure activity relationship (SAR) for this set follows the order: 7-OMe > 6-OMe > H > 7-Me > 6-Me.
Anti-Helicobacter pylori assay
The antibacterial potential of the hybrid molecules 4a-e was examined against three Helicobacter pylori strains resistant to metronidazole using the standard disk diffusion approach. The average inhibition zone diameters (IZDs) observed at varying concentrations (100, 50, 25, and 12.5 µg per disk) for the three isolates are presented in Table 2. According to the accepted evaluation guidelines, antibacterial activity was categorized as high (>20 mm), moderate (16-20 mm), weak (11-15 mm), or negligible (<10 mm).31
Table (2): Inhibition zone diameters of compounds 4a-e
 |
|||||
|---|---|---|---|---|---|
| Comp. | R | Average of inhibition zone diameters (range, mm)* | |||
| 100 µg/disk | 50 µg/disk | 25 µg/disk | 12.5 µg/disk | ||
| 4a | H | 22 (20-24) | 17 (21-23) | 15 (11-19) | 9 (7-11) |
| 4b | 6-Me | 11 (10-12) | 10 (8-11) | 7 (6-9) | 7 (5-9) |
| 4c | 6-OMe | 32 (30-34) | 26 (23-29) | 24 (20-27) | 21 (18-23) |
| 4d | 7-Me | 15 (12-19) | 12 (10-13) | 10 (8-12) | 8 (6-10) |
| 4e | 7-OMe | 38 (36-39) | 34 (32-35) | 30 (28-31) | 27 (24-29) |
| Metronidazole | – | 26 (24-31) | 24 (20-31) | 18 (14-21) | 12 (8-15) |
As shown in Table 2, most of the synthesized hybrids demonstrated notable growth inhibition against H. pylori within the concentration range of 100-25 µg/disk. Interestingly, their antibacterial performance corresponded closely to their urease inhibitory behavior. Compound 4e (R = 7-OMe), identified earlier as the most active urease inhibitor, also exhibited the strongest antibacterial response, producing inhibition zones exceeding 26 mm across all concentrations. It surpassed the reference antibiotic metronidazole, which showed a 26 mm zone at 100 µg/disk. The next most potent derivative, 4c (R = 6-OMe), generated inhibition zones between 21 mm at 12.5 µg/disk and 32 mm at 100 µg/disk. At every concentration, compound 4c proved superior to metronidazole approximately 1.5-fold stronger at 100, 50, and 25 µg/disk, and nearly 2.3-fold more active at 12.5 µg/disk. These findings highlight the dual bioactivity of the methoxy-bearing analogues 4c and 4e, confirming their role as both efficient urease inhibitors and potent anti-H. pylori agents.
The unsubstituted compound 4a (R = H) displayed substantial antibacterial activity at the highest dose (22 mm IZD at 100 µg/disk). However, its efficacy gradually declined to moderate (17 mm) and weak (15 mm) levels at 50 and 25 µg/disk, respectively, and it exhibited no measurable activity at 12.5 µg/disk. In contrast, the methyl-substituted counterparts 4b (R = 6-Me) and 4d (R = 7-Me) consistently showed only mild inhibitory effects throughout all tested concentrations, indicating the diminished antibacterial impact of methyl substitution compared to methoxy analogues.
Briefly, the synthesised hybrids demonstrated significant potential for dual action. In the urease inhibition assay, compounds 4c (IC50 = 0.75 µM) and 4e (IC50 = 0.58 µM) shown markedly superior potency relative to the reference Thiourea (IC50 = 21.50 µM). This sub-micromolar activity is comparable to or superior to that of previously documented benzimidazole and thiosemicarbazide derivatives, which typically exhibit IC50 values ranging from 1 to 10 µM.32,33 Moreover, compound 4e exhibited an inhibitory zone of 38 mm (at 100 µg/disk), significantly above the clinical standard Metronidazole, which measured 26 mm. At the lowest tested concentration (12.5 µg/disk), 4e (27 mm) demonstrated greater efficacy in inhibiting growth compared to Metronidazole at its maximum concentration (100 µg/disk, 26 mm). The methoxy-substituted hybrids exhibit superior enzyme inhibition and enhanced membrane permeability or intrinsic toxicity against H. pylori compared to standard nitroimidazole-based therapies.
This study systematically evaluated a designed series of quinoline-2-one/chalcone hybrid molecules (4a-e) to explore their dual pharmacological profile as urease inhibitors and anti-Helicobacter pylori agents. Biochemical assays confirmed that most of these synthesized hybrids exhibit strong urease inhibition, presenting IC50 values within the range of 0.58 µM to 1.90 µM. Their activity was markedly superior to that of the reference standard, thiourea (IC50 = 21.50 µM). Within the set, compounds 4a, 4c, and 4e stood out as the most active members. A notable and direct correlation was observed between urease inhibitory potential and antibacterial efficiency, as these same derivatives displayed pronounced inhibitory effects against H. pylori at concentrations from 100 down to 25 µg/disk. Collectively, these findings reinforce the potential of the quinoline-2-one/chalcone hybrid framework as an attractive and versatile scaffold for developing future anti-H. pylori therapeutics. Further investigations focusing on systematic modification and structure activity optimization of this molecular template are justified and may lead to promising lead compounds for subsequent preclinical evaluation.
Future perspectives
The remarkable efficacy of these hybrids, particularly compound 4e, highlights their potential as leading candidates for the treatment of H. pylori infections. Subsequent investigations will focus on testing the cytotoxicity of these compounds in mammalian cells, utilising established cell lines such as Vero or HEK-293, to further examine their translational significance and clinical safety. To ascertain the safety of these sub-micromolar urease inhibitors for humans, it is essential to determine the Selectivity Index (SI), defined as the ratio of the cytotoxic concentration (CC50) to the inhibitory concentration (IC50).
Additional file: Supplementary information.
ACKNOWLEDGMENTS
The author sincerely extends gratitude to Professor Dr. Bahaa G. M. Youssif, Department of Pharmaceutical Organic Chemistry, Faculty of Pharmacy, Assiut University, Egypt, for generously providing the synthesized compounds (4a–e) essential for the biological testing in this work. His valuable guidance and constructive input during the early preparation of the manuscript are deeply appreciated.
FUNDING
None.
DATA AVAILABILITY
All datasets generated or analyzed during this study are included in the manuscript and/or in the supplementary files.
ETHICS STATEMENT
Not applicable.
- Callahan BP, Yuan Y, Wolfenden R. The burden borne by urease. J Am Chem Soc. 2005;127(31):10828-10829.
Crossref - Ojha A, Bandyopadhyay TK, Das D. A comprehensive review on microbial urease: features and industrial applications. Crit Rev Biotechnol. 2026;46(1):1-24.
Crossref - Martinho N, Aniceto N. Decoding Urease Inhibition: A Comprehensive Review of Inhibitor Scaffolds. ChemMedChem 2026;21(2):e202500423.
Crossref - Asokan S, Banerjee N, Saleem M, et al. Healthcare associated infections (HAI): insights into epidemiology, microbiology, and diagnostics. Diagn Microbiol Infect Dis. 2026;15(3):117376.
Crossref - Almarmouri C, El-Gamal MI, Haider M, et al. Anti-urease therapy: a targeted approach to mitigating antibiotic resistance in Helicobacter pylori while preserving the gut microflora. Gut Pathogens. 2025;17(1):37.
Crossref - Guarner J, Mohar A, Parsonnet J, Halperin D. The association of Helicobacter pylori with gastric cancer and preneoplastic gastric lesions in Chiapas, Mexico. Cancer. 1993(71)2:297-301.
Crossref - Harris P, Mobley H, Perez-Perez G, Blaser M, Smith P. Helicobacter pylori urease is a potent stimulus of mononuclear phagocyte activation and inflammatory cytokine production. Gastroenterology. 1996;111(2):419-425.
Crossref - Covacci A, Telford JL, Giudice GD, Parsonnet J, Rappuoli R. Helicobacter pylori virulence and genetic geography. Science. 1999;284(5418):1328-1333.
Crossref - Montecucco C, Rappuoli R. Living dangerously: how Helicobacter pylori survives in the human stomach. Nat Rev Mol Cell Biol. 2001;2(6):457-466.
Crossref - Eaton KA, Brooks C, Morgan D, Krakowka S. Essential role of urease in pathogenesis of gastritis induced by Helicobacter pylori in gnotobiotic piglets. Infect Immun. 1991;59(7):2470-2475.
Crossref - Asokan S, Pandey RK, Jalil MA, et al. Biofilm associated infections on medical devices: pathogenesis, diagnostic challenges, and control strategies. The Microbe. 2026;11:100712.
Crossref - Levine C, Hiasa H, Marians KJ. DNA gyrase and topoisomerase IV: biochemical activities, physiological roles during chromosome replication, and drug sensitivities. Biochim Biophys Acta Gene Struct Expr. 1998;1400(1):29-43.
Crossref - Collin F, Karkare S, Maxwell A. Exploiting bacterial DNA gyrase as a drug target: current state and perspectives. Appl Microbiol Biotechnol. 2011;92(3):479-497.
Crossref - Cinelli MA, Mata SA, Watson T. Tropanes in natural products, syntheses, and drugs: some insights from the new millennium. Med Chem Res. 2026;35:686-710.
Crossref - Azimi SG, Shakour N, Bagherzade G, Saberi MR, Azimi H, Moosavi FM. A comprehensive review of the biological activities of medicinal metal complexes synthesized from Quinoline scaffolds. Bioinorg Chem Appl. 2025;2025(1):3133615.
Crossref - Elshaier YA, Aly AA, Abdel-Aziz M, Fathy HM, Brown AB, Brase S, Ramadan M. Synthesis and Identification of New N, N-Disubstituted Thiourea, and Thiazolidinone Scaffolds Based on Quinolone Moiety as Urease Inhibitor. Molecules. 2022;27(20):7126.
Crossref - Elbastawesy MA, Aly AA, El-Shaier YA, Brown AB, Abuo-Rahma GE-DA, Ramadan M. New 4-thiazolidinone/quinoline-2-ones scaffold: Design, synthesis, docking studies and biological evaluation as potential urease inhibitors. J Mol Struct. 2021;1244:130845.
Crossref - Patil P, Khan P, Zangade S. Synthesis of 1, 3-diaryl-2-propene-1-one derivatives using Tripotassium phosphate as an alternative and efficient catalyst and study its cytotoxic and antimicrobial properties. Curr Chem Lett. 2020;9(4):183-198.
Crossref - Zhuang C, Zhang W, Sheng C, Zhang W, Xing C, Miao Z. Chalcone: a privileged structure in medicinal chemistry. Chem Rev. 2017;117(12):7762-7810.
Crossref - Alidmat M, Alshhab A, Alokour M. Chalcones: Multifunctional Scaffolds Bridging Synthesis, Structure-Activity Relationships, and Therapeutic Applications: A Review. J Chem Rev. 2026;8(2):296-321.
Crossref - Abou-Zied HA, Youssif BG, Mohamed MF, Hayallah AM, Abdel-Aziz M. EGFR inhibitors and apoptotic inducers: Design, synthesis, anticancer activity and docking studies of novel xanthine derivatives carrying chalcone moiety as hybrid molecules. Bioorganic Chem. 2019;89:102997.
Crossref - Hisham M, Hassan HA, Gomaa HA, Youssif BG, Hayallah AM, Abdel-Aziz M. Structure-based design, synthesis and antiproliferative action of new quinazoline-4-one/chalcone hybrids as EGFR inhibitors. J Mol Struct. 2022;1254:132422.
Crossref - Akter R, Islam MM, Islam MW, et al. Recent Developments in the Biological Consequences of Certain Chalcone Derivatives. ChemistrySelect. 2025;10(41):e03482.
Crossref - Kaoud TS, Mohassab AM, Hassan HA, et al. NO-releasing STAT3 inhibitors suppress BRAF-mutant melanoma growth. Eur J Med Chem. 2020;186:111885.
Crossref - Mohassab AM, Hassan HA, Abdelhamid D, et al. Design and synthesis of novel quinoline/chalcone/1, 2, 4-triazole hybrids as potent antiproliferative agent targeting EGFR and BRAFV600E kinases. Bioorg Chem. 2021;106:104510.
Crossref - Abdelbaset MS, Abdelrahman MH, Bukhari SNA, et al. Design, synthesis, and biological evaluation of new series of pyrrol-2(3H)-one and pyridazin-3(2H)-one derivatives as tubulin polymerization inhibitors. Bioorg Chem. 2021;107:104522.
Crossref - Adhikari S, Nath P, Deb VK, et al. Pharmacological potential of natural chalcones: A recent studies and future perspective. Front Pharmacol. 2025;16:1570385.
Crossref - Youssif B. Synthesis and biological evaluation of novel quinoline/chalcone hybrid as potential antibacterial agents. Int J Pharm Sci Res. 2019;10:2423-2429.
Crossref - Pervez H, Iqbal MS, Tahir MY, Nasim F-u-H, Choudhary MI, Khan KM. In vitro cytotoxic, antibacterial, antifungal and urease inhibitory activities of some N 4-substituted isatin-3-thiosemicarbazones. J Enzyme Inhib Med Chem. 2008;23(6):848-854.
Crossref - Ibrahim TS, Bokhtia RM, Al-Mahmoudy AM, et al. Design, synthesis and biological evaluation of novel 5-((substituted quinolin-3-yl/1-naphthyl) methylene)-3-substituted imidazolidin-2, 4-dione as HIV-1 fusion inhibitors. Bioorg Chem. 2020;99:103782.
Crossref - Moshafi MH, Sorkhi M, Emami S, et al. 5-Nitroimidazole-based 1,3,4-Thiadiazoles: Heterocyclic Analogs of Metronidazole as Anti-Helicobacter pylori Agents. Archiv der Pharmazie. 2011;344(3):178-183.
Crossref - Alzahrani AYA, Adalat B, Ullah H, et al Design, synthesis, in vitro urease inhibitory potentials and in silico molecular docking study of benzimidazole bearing thiosemicarbazides/sulfonamide Analogues. J Mol Struct. 2024;1296(part 2):136850.
Crossref - Ali B, Khan KM, Arshia, et al. Synthetic nicotinic/isonicotinic thiosemicarbazides: In vitro urease inhibitory activities and molecular docking studies. Bioorg Chem. 2018;79:34-45.
Crossref
© The Author(s) 2026. Open Access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License which permits unrestricted use, sharing, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.