


ISSN: 0973-7510
E-ISSN: 2581-690X
The study focused on developing an indigenous, one-step reverse transcription (RT)-PCR assay for detecting the porcine transmissible gastroenteritis virus (TGEV) genome. In summary, a gene construct and two sets of primers were designed by aligning N gene sequences from various TGEV strains, which were subsequently synthesized. The gene construct was sub-cloned into the pTZ57R/T vector, enabling the synthesis of in vitro transcribed (IVT) RNA, which served as a TGEV-positive control for RT-PCR protocol optimization. The assay optimization involved systematic testing of various parameters, including primer concentrations, magnesium (Mg++) levels, RNA template quantities, annealing temperatures, and other thermal variables. The analytical sensitivity was evaluated by examining serial 10-fold dilutions of IVT-RNA, both in actual form and when recovered from swine feces after spiking with the same dilutions of IVT-RNA. The developed assay demonstrated analytical sensitivities of 47.548 × 10² and 24.629 × 10³ RNA copies at 10-7 and 10-6 dilutions of IVT-RNA and spiked fecal RNA, respectively. Specificity was confirmed by testing against porcine epidemic diarrhea virus (PEDV), porcine reproductive and respiratory syndrome virus (PRRSV), classical swine fever virus (CSFV), swine influenza virus (SIV), and known TGEV-negative swine fecal or rectal swab samples (n = 320) collected from the field. The assay exhibited specific amplification for TGEV without cross-reactivity to PEDV, PRRSV, CSFV, SIV, or field samples. This one-step RT-PCR assay proved to be both sensitive and specific for TGEV genomic detection, offering a reliable diagnostic tool for future outbreaks and subsequent monitoring of TGE.
Diagnostic Preparedness, Disease Monitoring, Genomic Detection, Pigs, Reverse Transcription-PCR, Transmissible Gastroenteritis, TGE Virus
Transmissible gastroenteritis (TGE) is a highly infectious and contagious viral disease affecting the gastrointestinal system of pigs, caused by the TGE virus (TGEV), which belongs to the genus Alphacoronavirus within the family Coronaviridae.1,2 The disease is marked by severe watery diarrhea, vomiting, dehydration, and significant case fatality, particularly in neonatal piglets under two weeks of age, where mortality rates can reach as high as 100%.3
TGE was initially documented in the United States in 1946.3 Since then, it has spread to almost all the continents except Australia and Antarctica. In Asia, cases of TGE have been reported in countries such as China,4,5 Japan,6 and South Korea.7 Notably, epidemics in China have unveiled the emergence of novel TGEV strains that bear a close genetic resemblance to strains found in the United States, highlighting the ability of TGEV to undergo mutations.8,9 Some recombinant TGEV strains have been shown to experimentally infect multiple host species, adding complexity to both diagnosis and control measures.10
The economic impact of TGE on the swine industry is substantial and is directly attributed to high mortality rates and production losses among pigs.11 Additional economic burdens arise from expenses related to vaccination and biosecurity measures. For instance, in Australia, a study estimated economic losses ranging from $260 to $330 per breeding sow in the year following TGEV infection.12 Considering its impact on food security and animal health, the global eradication of TGE should remain the ultimate goal.
Although serological evidence of TGE has been documented in the Indian states of Assam13 and Uttar Pradesh,14 the presence of TGEV or clinical cases of the disease in Indian pig populations has not yet been reported. Nevertheless, the proximity of clinical TGE cases in China and some Southeast Asian countries increases the likelihood of its emergence in India. Due to the importance of swine production in many regions, preventing the introduction of TGE across borders is of utmost importance. Rapid and accurate diagnostic capabilities are essential for the timely implementation of effective control measures against the disease. To cater to this need, the current study was undertaken to develop a suitable RT-PCR assay for the genomic detection of TGEV in pigs, ensuring diagnostic preparedness against TGE.
Designing of primers and synthetic gene construct
The nucleoprotein (N) gene sequences of various transmissible gastroenteritis virus (TGEV) strains, available in the online database (NCBI), were aligned to design primers and a synthetic gene construct. A conserved region of the N gene, present across multiple TGEV strains, was identified and used to design suitable primers through the online Primer3 (v. 0.4.0) software tool.15,16 To ensure broader coverage, degenerate nucleotides were incorporated into the primer sequences. Two sets of primers were designed: Set 1 included 5’-CTAGAGGCAGGCAACAATYC–3’ as a forward primer and 5’-CRAGGYCACTGTCACCAAAA-3’ as a reverse primer, yielding an expected amplicon of 267 bp. Set 2 comprised a forward primer 5’-TCAGCCAATTTTGGTGACA-3’ and a reverse primer 5’-GATGGRCGAGCATAGGCATT-3’, expected to amplify a 233 bp amplicon. A 650 bp fragment of the TGEV N gene, positions 500 to 1149, was selected for the construct and synthesized commercially within the pUC57 vector (Eurofins Genomics India Pvt. Ltd., India).
Sub-cloning of the gene construct
The TGEV N gene construct was excised from the pUC57 vector through restriction endonuclease (RE) digestion using EcoRI and HindIII enzymes. The released fragment was subsequently gel-purified and ligated into the pTZ57R/T vector (Thermo Scientific, USA). The ligated vector was transformed into competent Escherichia coli JM109 cells, which were then cultured overnight on Luria-Bertani (LB) agar plates in the presence of ampicillin at a concentration of 100 µg/mL at 37 °C. Recombinant colonies, identifiable by their white coloration, were screened for the presence of the target gene insert by PCR and cultured overnight in LB broth containing ampicillin (100 µg/mL) at 37 °C under constant shaking. Plasmid DNA was extracted from the broth culture of the confirmed recombinant colonies using a plasmid extraction kit (Promega Corporation, USA) and subsequently linearized through digestion by EcoRI enzyme.
In vitro transcription (IVT) of RNA
The linearized DNA was precipitated using 0.5 M EDTA and 3 M sodium acetate in absolute ethanol. Following proteinase K treatment, the precipitated DNA was purified using the Phenol-chloroform method.17 Before proceeding with IVT-RNA synthesis, the purified DNA was screened for the presence of the TGEV N gene through PCR analysis.
The IVT process was carried out using the linearized plasmid containing the TGEV N gene and T7 RNA polymerase, utilizing the T7 transcription kit (Invitrogen, USA) as per a previously established method.18 To eliminate residual DNA, the transcribed RNA was treated with 8 U of TURBO™ DNase (Invitrogen, USA) at 37 °C for 75 min. The DNase enzyme was subsequently inactivated through ammonium acetate treatment. The RNA, now free of residual DNA, was purified using the Phenol-chloroform method.17 The purified RNA precipitate was air-dried, dissolved in 20 µL of nuclease-free water (NFW), and kept at -80 °C for further use.
Testing of IVT-RNA
The synthesized IVT-RNA was evaluated using one-step RT-PCR to confirm successful transcription and PCR to verify the absence of residual DNA. The RT-PCR product was analyzed by gel electrophoresis. The amplified product was extracted from the gel using the gel purification kit (Qiagen, Germany) and subsequently subjected to nucleotide sequencing using Sanger’s sequencing method for confirmation.
Quantification of IVT-RNA
The concentration of IVT-RNA was estimated using Qubit 4 Fluorometer (Thermo Scientific, USA) and expressed as copy numbers. The IVT-RNA was serially diluted (10-fold) in NFW for use in fecal spiking and to evaluate analytical sensitivity of RT-PCR assay.
Optimization of RT-PCR protocol
The RT-PCR protocol was initially optimized with varying parameters such as primer concentrations (0.2, 0.4, and 0.6 µM), Mg++ concentration, RNA template (1 to 5 µl of 1:10000 diluted IVT-RNA), annealing temperatures (47 to 57 °C) and other thermal cycling conditions (Table 1). Each run included TGEV N gene-specific IVT-RNA as the positive control and a no-template control (NTC). The amplified products, along with a suitable DNA marker, were evaluated through gel electrophoresis screening.
Table (1):
Thermal conditions used for RT-PCR optimization
Thermal cyclic parameters |
Temp. (°C) |
Cycling time |
Cycle (n) |
|---|---|---|---|
cDNA synthesis |
45 |
30 min |
01 |
Initial denaturation |
95 |
10/5/3 min |
01 |
Cyclic denaturation |
95 |
30/20 sec |
35/39 |
Annealing of primers |
47-57 |
30 sec |
35/39 |
Elongation |
72 |
45/30 sec |
35/39 |
Final elongation |
72 |
7/5 min |
01 |
Fecal spiking with IVT-RNA
The assay was further evaluated by amplifying RNA recovered from fecal samples spiked with IVT-RNA. For the spiking process, 20% (w/v) homogenates of swine feces, previously tested and confirmed negative for the TGEV genome, were prepared in sterile phosphate-buffered saline (PBS) and subsequently clarified by low-speed centrifugation (1000 g). From the clarified homogenate, 140 µL was spiked with 10 µL of each IVT-RNA dilution. RNA was then extracted from these spiked fecal samples using a viral RNA extraction kit (Qiagen, Germany) and eluted in 35 µL of the provided elution buffer. The extracted RNA was subsequently analyzed for the presence of the TGEV N gene using the optimized RT-PCR protocol.
Evaluation of analytical sensitivity and specificity
The performance of the optimized assay was evaluated by determining both analytical sensitivity and specificity. Analytical sensitivity was estimated in serially diluted IVT-RNA and RNA extracted from spiked fecal homogenates by calculating the copy number at the endpoint dilution, where a distinct amplification band was visible. The RNA copies were calculated using the following standard formulas:
Initially, the number of moles of single-stranded (ss) RNA was estimated as per the following formula:
Moles of ssRNA are equal to mass of ssRNA (g)/molecular weight of ssRNA (g/mol).
The molecular weight of ssRNA = (Numbers of ssRNA nucleotides × average molecular weight of an ssRNA nucleotide) + 18.02 g/mol. (The average molecular weight of an ssRNA nucleotide is around 340 g/mol for an unmodified nucleotide. However, when including the phosphate group as part of a ribonucleotide monophosphate, the average molecular weight is approximately 321.47 g/mol, which is often used in ssRNA molecular weight calculations).
The number of ssRNA molecules (copy number) was then derived by multiplying the moles of ssRNA by Avogadro’s number (6.02214 × 1023 molecules/mol).
The analytical specificity of the optimized assay was evaluated using RNA templates specific to various swine viruses, including porcine epidemic diarrhea virus (PEDV), porcine reproductive and respiratory syndrome virus (PRRSV) genotypes 1 (EU) and 2 (NA), classical swine fever virus (CSFV) and swine influenza virus (SIV). To further verify specificity, the assay was tested on 320 swine fecal samples collected from the field, which were known to be negative (these negative samples were confirmed through three blind passages in PK-15 cells, followed by testing with TGE-specific RT-PCR).19
The PCR amplification using recombinant plasmid DNA as a template resulted in the expected amplicon (267 bp), confirming the presence of the TGEV N gene insert in the pTZ57R/T vector. Based on the PCR amplification results from the initial optimization process with two primer sets, primer set-1 was selected for further optimization of the RT-PCR assay (Figure 1A). Primer set-2 was excluded from further optimization due to non-specific amplification.
The successful amplification observed using the RT-PCR protocol confirmed the correct synthesis of the IVT-RNA, and the lack of amplification using the PCR protocol indicated the absence of residual DNA in the IVT-RNA (Figure 1B). Sanger’s sequencing of the RT-PCR product further validated the specificity of TGEV N gene amplification.
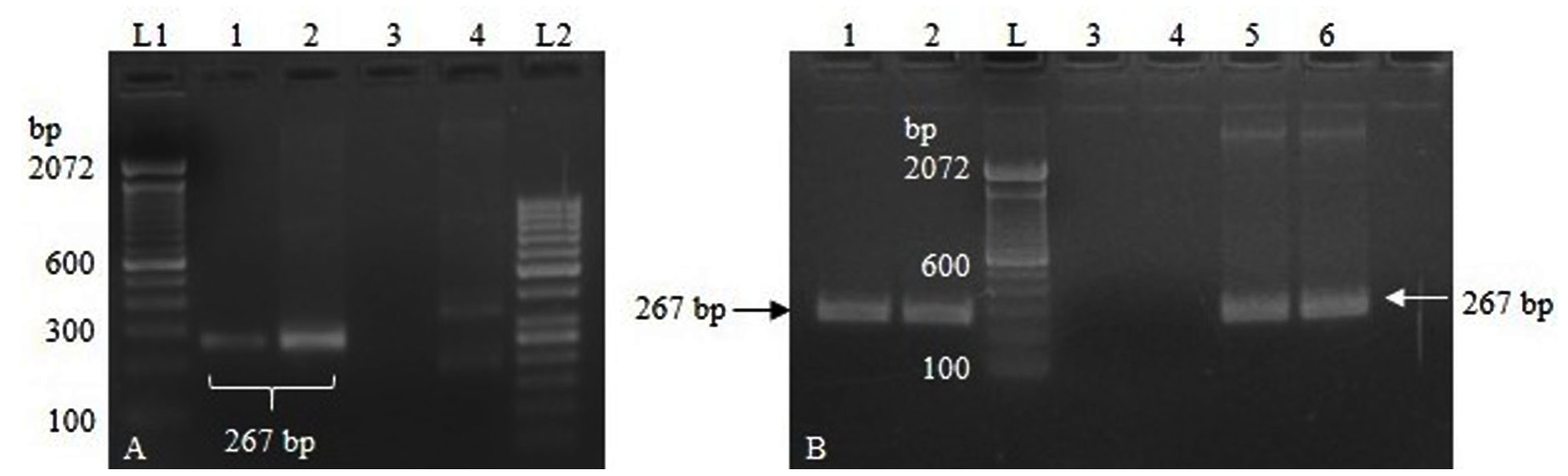
Figure 1. (A). Lane 1-2: PCR amplification using primer set 1. Lane 3-4: PCR amplification using primer set 2. Lane L1, L2: DNA ladder; (B). Lane 1-2: RT-PCR using IVT-RNA template showing amplification specific to TGEV N gene. Lane 3-4: PCR showing no amplification using IVT-RNA template, indicating complete depletion of residual DNA. Lane 5-6: PCR amplification using linearized DNA as a template. Lane L: DNA ladder
The concentration of the IVT-RNA was estimated to be 8.25 µg/mL, corresponding to 39.478 femtomoles (39.478 × 10-15 moles), or 23.774 × 109 RNA copies per µL. The RT-PCR reactions demonstrated visible amplification bands up to dilutions of 10-6 and 10-7 of the IVT-RNA template after 35 and 39 cycles, respectively (Figure 2). After spiking, a clear amplification band was observed at spiking dilutions up to 10-6 following 39 cycles (Figure 3).
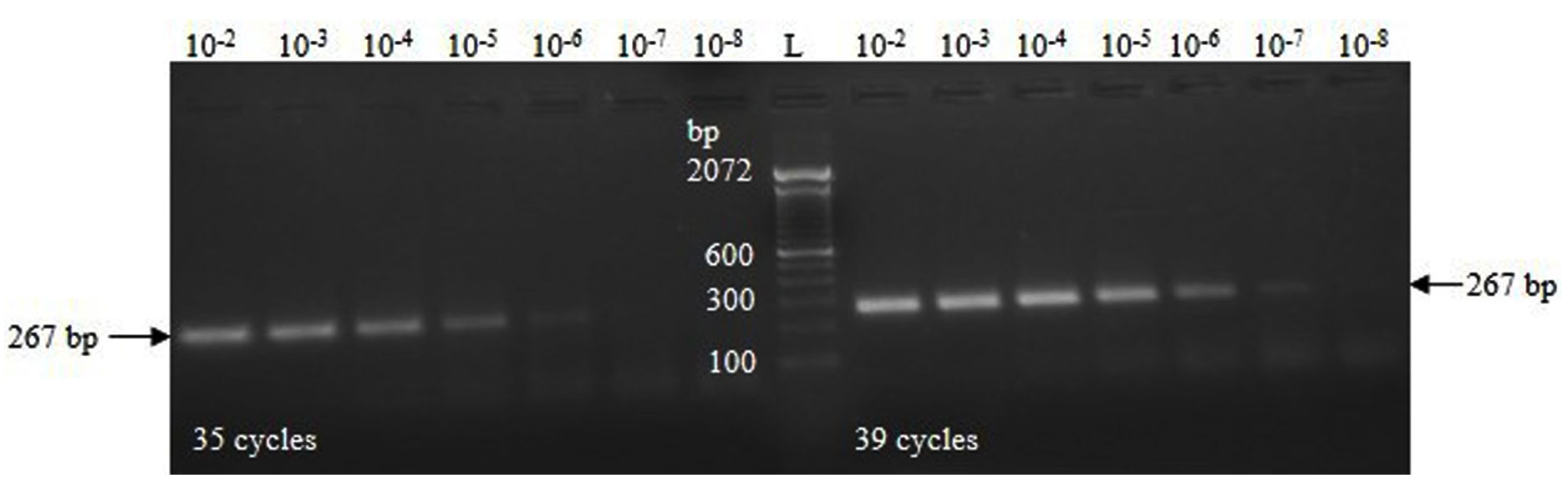
Figure 2. RT-PCR amplification using serial dilutions (10-fold) of the IVT-RNA specific to the TGEV N gene
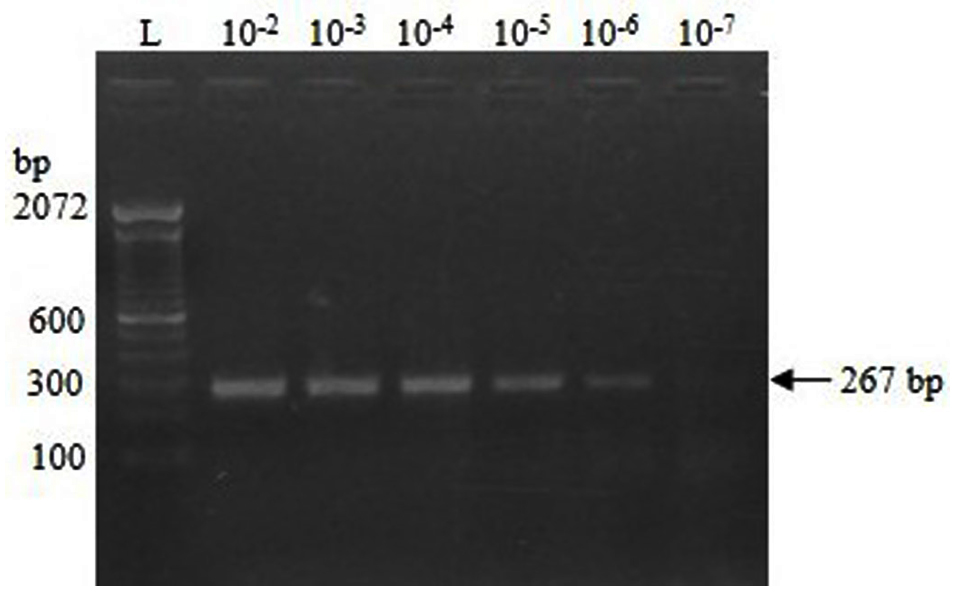
Figure 3. RT-PCR amplification of RNA extracted from fecal samples spiked with serial 10-fold dilutions of IVT-RNA
The final optimized one-step RT-PCR reaction mixture comprised 4 µL of reaction buffer (5×), 0.2 mM concentration of dNTPs, 0.75 mM concentration of magnesium sulfate, 2 U of reverse transcriptase enzyme, 2 U of DNA polymerase enzyme, 0.4 µM concentration of each primer, 2 µL of RNA template, and finally NFW bringing the total volume to 20 µL.
The optimized cycling parameters were as follows: RNA reverse transcription at 45 °C for 30 min followed by an initial denaturation cycle of 3 min at 95 °C. This was followed by 39 amplification cycles, which included a denaturation step at 95 °C for 20 sec, an annealing step at 52 °C for 30 sec, and an elongation step at 72 °C for 30 sec. A final elongation step was performed at 72 °C for 5 min. The protocol, including the cDNA synthesis step, was completed in 1 hour and 45 min.
The RT-PCR assay exhibited a clear amplification at a 10-7 dilution of the IVT-RNA template (2 µL), with an analytical sensitivity of 47.548 × 10² RNA copies (2 × 23.774 × 109 × 10-7). After spiking fecal samples with IVT-RNA, the assay demonstrated an analytical sensitivity of 24.629 × 10³ RNA copies (2 × 23.774 × 109 × 10-6 × 0.518) at a spiking dilution of 10-6. In this context, the multiplication factor of 0.518 is derived from the value of approximately 10-0.286, corresponding to the dilution of 10 µL of IVT-RNA eluted in a total volume of 35 µL. The RNA copy numbers in various dilutions of IVT-RNA and RNA extracted following fecal spiking are presented in Table 2.
Table (2):
Number of RNA copies in different dilutions of the IVT-RNA and in RNA recovered after faecal spiking
| Serial dilutions | Synthesized IVT-RNA | RNA from spiked faeces |
|---|---|---|
| RNA copies per reaction | RNA copies per reaction | |
| Undiluted | 47.548 × 109 | 24.629 × 109 |
| 10˗1 | 47.548 × 108 | 24.629 × 108 |
| 10˗2 | 47.548 × 107 | 24.629 × 107 |
| 10˗3 | 47.548 × 106 | 24.629 × 106 |
| 10˗4 | 47.548 × 105 | 24.629 × 105 |
| 10˗5 | 47.548 × 104 | 24.629 × 104 |
| 10˗6 | 47.548 × 103 | 24.629 × 103* |
| 10˗7 | 47.548 × 102* | 24.629 × 102 |
| 10˗8 | 47.548 x 101 | 24.629 × 101 |
*detection limit
The RT-PCR assay was found to be highly specific for detecting the TGEV genome, because there was no amplification with RNA from PEDV, PRRSV, CSFV, or SIV. Moreover, all 320 known negative field samples tested were consistently detected as negative, further validating the specificity of the assay.
TGE is a transboundary disease capable of spreading across vast geographical areas, including continents. It is recognized as a reportable disease by the WOAH. Transmission of TGEV primarily occurs through a fecal-oral route, with infected pigs excreting large quantities of the virus in their feces. This makes feces a major source of TGEV transmission, either directly through carrier pigs or indirectly through some mechanical modes.2 TGE outbreaks are common in immunologically naive herds.20 Due to the occurrence of TGE in neighboring countries like China and the frequent movement of humans and animals across international borders, especially in India’s northeastern states, there is a persistent risk of the disease spreading into India.
Although no antigenic or genomic evidence of TGEV has been reported in India, earlier studies have documented the prevalence of TGEV antibodies in 20.11% and 39.4% of pig populations in Uttar Pradesh and Assam, respectively.13,14 This highlights the importance of active surveillance and regular monitoring of the Indian pig population using specific diagnostic tools, such as RT-PCR, to gain a clearer understanding of the current status of TGEV in the country. Effective control and prevention strategies for TGE rely on rapid and accurate diagnosis, stringent biosecurity measures, and the maintenance of herd immunity on farms.21
While TGE can be presumptively diagnosed based on the history, clinical signs, and pathological findings at the necropsy, its clinical signature in endemic cases is not distinctive enough for a definitive diagnosis. Endemic TGE often appears in a mild, chronic form, with intermittent episodes of diarrhea in suckling or weaned piglets, making it challenging to diagnose.2,20 Moreover, the emergence and co-infection with other enteric viral pathogens, such as rotavirus A and C, other porcine coronaviruses, including PEDV, swine acute diarrhea syndrome coronavirus, porcine delta coronavirus, and the porcine hemagglutinating encephalomyelitis virus (which causes vomiting and wasting disease), lead to similar clinical symptoms, complicating diagnosis.22-24 Therefore, TGE must be differentiated from these diseases for a specific and accurate diagnosis. Furthermore, it should also be distinguished from non-viral infections caused by several bacterial pathogens, such as Escherichia coli, Clostridium spp., Salmonella spp., etc., as well as many parasitic pathogens like certain nematodes, Cystoisospora sp., etc., all of which can be present with similar clinical signs.20,25
Hence, laboratory testing using one or more specific diagnostic techniques, such as PCR is required to perform an accurate disease diagnosis.21 PCR-based techniques are highly useful for detecting the genome of the target pathogen in a variety of clinical or biological specimens such as feces, intestinal samples,
etc.26,27 For RNA viruses like PEDV or TGEV, RT-PCR technique can be applied to test the weaned and older pigs, which have low virus load, exhibit mild clinical signs, or possess less prominent lesions.28,29 Several one-step RT-PCR assays have previously been employed for the genomic testing of TGEV.19,29
To assess the performance of a diagnostic test, specificity, and sensitivity are considered the most important parameters. Various factors, including the primer sequences, play a significant role in determining the specificity and sensitivity of the assays meant for genomic amplification purposes.19,30,31 The use of primers with degenerate nucleotides in the present RT-PCR assay ensures broader coverage of the target sequence, which may enhance both diagnostic specificity and sensitivity. Additionally, the ability of this RT-PCR assay to detect low copy numbers of the TGEV N gene makes it a useful tool for the accurate diagnosis of TGE.
Though the viruses can be detected by several other techniques like fluorescent antibody tests, antigen-detecting ELISAs, streptavidin-biotin methods, etc., these techniques are cumbersome and require more time to perform.19 The present RT-PCR assay is rapid to perform and can give results quickly using nucleic acid from clinical fecal samples or rectal swabs of the pigs. TGE diagnostic facilities using conventional PCR can be easily established in Veterinary healthcare institutions and peripheral diagnostic laboratories.
This study presents a highly specific and sensitive one-step RT-PCR assay designed to detect the TGEV genome in fecal samples from pigs. The assay holds the potential for diagnosing TGE in case of future outbreaks and can also be employed for further monitoring of TGE in the Indian pig population, aiding the implementation of suitable and effective control strategies against the disease.
ACKNOWLEDGMENTS
The authors thank the Director of ICAR-National Institute of High Security Animal Diseases, Bhopal, and the Indian Council of Agricultural Research (ICAR), New Delhi, for providing financial support under institutional research project (Project code IXX 13779) to carry out the present research work.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
AUTHORS’ CONTRIBUTION
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
FUNDING
None.
DATA AVAILABILITY
All datasets generated or analysed during this study are included in the manuscript.
ETHICS STATEMENT
Not applicable.
- Gonzalez JM, Gomez-Puertas P, Cavanagh D, Gorbalenya AE, Enjuanes L. A comparative sequence analysis to revise the current taxonomy of the family Coronaviridae. Arch Virol. 2003;148(11):2207-2235.
Crossref - Saif LJ. Transmissible gastroenteritis. Chapter 3.8.10. OIE Terrestrial Manual. 2018:1627-1638.
- Doyle LP, Hutchings LM. A transmissible gastroenteritis in pigs. J Am Vet Med Assoc. 1946;108:257-259.
- Zhenhui S, Xianjin D, Xinzhi C, Yue Z, Yuntian L, Yong L. Isolation and Identification of a New Strain of Porcine Transmissible Gastroenteritis Virus from Chongqing, Southwestern China. Isr J Vet Med. 2015;70(4): 22-30.
- Yuan D, Yan Z, Li M, Wang Y, Su M, Sun D. Isolation and Characterization of a Porcine Transmissible Gastroenteritis Coronavirus in Northeast China. Front Vet Sci. 2021;8:611721.
Crossref - Usami Y, Fukai K, Ichikawa Y, et al. Virological and serological studies of porcine respiratory coronavirus infection on a Japanese farm. J Vet Med Sci. 2008;70(9):929-936.
Crossref - Park JH, Han JH, Kwon HM. Sequence analysis of the ORF 7 region of transmissible gastroenteritis viruses isolated in Korea. Virus Genes. 2008;36(1):71-78.
Crossref - Hou Y, Yue X, Cai X, et al. Complete genome of transmissible gastroenteritis virus AYU strain isolated in Shanghai, China. J Virol. 2012;86(21):11935.
Crossref - Kanner-Acerbo E, Lowe J. Review of immunological responses to porcine coronaviruses and implications on population based control strategies in epidemic and endemic infections. World J Immunol2016; 6(1):60-66.
Crossref - Chen Y, Zhang Y, Wang X, et al. Transmissible Gastroenteritis Virus: An Update Review and Perspective. Viruses. 2023;15(2):359.
Crossref - Pritchard GC. Observations on clinical aspects of transmissible gastroenteritis of pigs in Norfolk and Suffolk, 1980-81. Vet Rec. 1982;110(20):465-469.
Crossref - Mullan BP, Davies GT, Cutler RS. Simulation of the economic impact of transmissible gastroenteritis on commercial pig production in Australia. Aust Vet J. 1994;71(5):151-154.
Crossref - Barman NN, Barman B, Sharma DK, Pensaert MB. Prevalence of Rotavirus, transmissible gastroenteritis virus and porcine epidemic diarrhoea virus antibodies in pigs of Assam, India. Indian J Ani Sci. 2003;73(6):576–578.
- Rout M, Saikumar G. Serological prevalence of transmissible gastroenteritis virus infection in swine. Indian Vet J. 2012;89(12):13-15.
- Koressaar T, Remm M. Enhancements and modifications of primer design program Primer3. Bioinformatics. 2007;23(10):1289-1291.
Crossref - Untergasser A, Cutcutache I, Koressaar T, et al. Primer3-new capabilities and interfaces. Nucleic Acids Res. 2012;40(15):e115.
Crossref - Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual, 3rd Ed. Vol. 1, Cold Spring Harbor Laboratory Press, New York. 2001.
- Milligan JF, Groebe DR, Witherell GW, Uhlenbeck OC. Oligoribonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates. Nucleic Acids Res. 1987;15(21):8783-8798.
Crossref - Kim SY, Song DS, Park BK. Differential detection of transmissible gastroenteritis virus and porcine epidemic diarrhea virus by duplex RT-PCR. J Vet Diagn Invest. 2001;13(6):516-520.
Crossref - Saif LJ, Pensaert MB, Sestak K, Yeo SG, Jung K. Coronaviruses. Diseases of Swine. 10th Ed. John Wiley & Sons, Inc, New York. 2012:501-524.
- Saif LJ, Sestak K. Transmissible gastroenteritis virus and porcine respiratory coronavirus. In: Straw BE, Zimmerman JJ, D’Allaire S, Taylor DJ, eds. Diseases of Swine. Ames, IA: Blackwell Publishing; 2006:489-516. ISBN 0-8138-1703-X.
- Marthaler D, Raymond L, Jiang Y, Collins J, Rossow K, Rovira A. Rapid detection, complete genome sequencing, and phylogenetic analysis of porcine deltacoronavirus. Emerg Infect Dis. 2014;20(8):1347-1350.
Crossref - Chen F, Knutson TP, Rossow S, Saif LJ, Marthaler DG. Decline of transmissible gastroenteritis virus and its complex evolutionary relationship with porcine respiratory coronavirus in the United States. Sci Rep. 2019;9(1):3953.
Crossref - Wang Q, Vlasova AN, Kenney SP, Saif LJ. Emerging and re-emerging coronaviruses in pigs. Curr Opin Virol. 2019;34:39-49.
Crossref - World Organization for Animal Health (OIE). Infection with porcine epidemic diarrhea virus. OIE Technical Factsheet. 2014:1-4. https://www.woah.org/fileadmin/Home/eng/Our_scientific_expertise/docs/pdf/A_factsheet_PEDV.pdf.
- Jung K, Saif LJ. Porcine epidemic diarrhea virus infection: Etiology, epidemiology, pathogenesis and immunoprophylaxis. Vet J. 2015;204(2):134-143.
Crossref - Diel DG, Lawson S, Okda F, et al. Porcine epidemic diarrhea virus: An overview of current virological and serological diagnostic methods. Virus Res. 2016;226:60-70.
Crossref - Olech M. Current State of Molecular and Serological Methods for Detection of Porcine Epidemic Diarrhea Virus. Pathogens. 2022;11(10):1074.
Crossref - Song DS, Kang BK, Oh JS, et al. Multiplex reverse transcription-PCR for rapid differential detection of porcine epidemic diarrhea virus, transmissible gastroenteritis virus, and porcine group A rotavirus. J Vet Diagn Invest. 2006;18(3):278-281.
Crossref - Erlich HA, Gelfand DH, Saiki RK. Specific DNA amplification. Nature. 1988;331(6155):461-462.
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual, 2nd Ed. Cold Spring Habor Laboratory Press, Cold Spring Habor, New York. 1989.
© The Author(s) 2025. Open Access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License which permits unrestricted use, sharing, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.