


ISSN: 0973-7510
E-ISSN: 2581-690X
Geothermal ecosystems are underexplored reservoirs of microbial diversity with significant biogeochemical and biotechnological implications. This study presents a high-resolution metagenomic analysis of thermophilic microbial communities in the hot mud of Linow Lake, a tropical geothermal system in Indonesia’s Wallacea Zone a region straddling the Pacific and Mediterranean volcanic cirques. Using 16S rRNA gene sequencing and co-occurrence network analysis, we identified a microbial community dominated by Proteobacteria (65% Burkholderiaceae, 24.6% Corynebacteriaceae), with novel, unclassified taxa (0.1%), highlighting the site’s unique biodiversity. Network analysis revealed tightly clustered functional guilds, suggesting metabolic syntrophy in sulfur cycling and organic matter degradation under extreme temperature (50-98 °C) and acidic (pH 2-4) conditions. Notably, the dominance of Burkholderia and Corynebacterium genera implies adaptive strategies for heavy metal resistance and bioactive compound synthesis. These findings underscore the ecosystem’s role in global biogeochemical cycles and its untapped potential for industrial applications, including thermostable enzyme production and bioremediation. Our work bridges critical gaps in understanding tropical geothermal microbiomes and positions Linow Lake as a key site for exploring microbial evolution and bioprospecting in multi-stress environments.
Thermophiles, Metagenomics, Co-occurrence Network, Linow Lake, Hot Mud
North Sulawesi, Indonesia, holds unique scientific significance as part of the Wallacea Zone a biogeographic transition region between Asia and Australasia and is crossed by two global volcanic cirques (the Pacific and Mediterranean).1,2 This combination of geological factors creates a geothermal ecosystem with extreme physicochemical characteristics, such as variations in temperature, pH, and mineral content not found in other regions.3 Transition zones like Wallacea tend to harbor endemic microbial communities with unique metabolic adaptations, including sulfur oxidation and heavy metal reduction capabilities.4 Metagenomic research in this area has the potential to uncover new bacterial taxa and genes encoding thermostable enzymes not yet recorded in global databases.4 Furthermore, the complex interactions between the two volcanic systems may trigger the evolution of distinct metabolic pathways, such as syntrophy between thermophilic microbes.5,6
The dominance of phyla such as Proteobacteria and Actinobacteria in the hot springs of North Sulawesi indicates their potential in heavy metal bioremediation and bioactive compound production.7 Furthermore, co-occurrence network analysis can reveal microbial functional guilds that play a role in stabilizing geothermal ecosystems.8 These findings not only contribute to mapping global microbial diversity but also open up opportunities for industrial applications, such as the development of thermostable enzymes for bioprocessing and biomining. Thus, metagenomic research in North Sulawesi plays an important role in exploring potentially untapped microbial resources in a highly active geodynamic region.
Geothermal ecosystems, such as Lake Linow in Indonesia, are extreme environments rich in unique microbial diversity with special adaptations to high temperatures, acidity, and extreme mineral content.9 Previous research by the authors found nine bacterial isolates with thermostable lipase activity from Lake Linow’s hot mud.9 The microbial communities in these environments play a key role in biogeochemical cycles, including sulfur, carbon, and heavy metal cycling, which impact overall ecosystem productivity.10 However, most previous geothermal research has focused on temperate locations such as Yellowstone or Iceland, while tropical geothermal ecosystems like Lake Linow remain underexplored.11,12 Metagenomic studies in this environment are not only important for understanding microbial ecology but also open up opportunities to discover thermostable enzymes and bioactive compounds with potential industrial applications.13
16S rRNA gene-based metagenomic analysis has become a standard approach for characterizing microbial communities in extreme environments.14 However, most studies focus solely on taxonomic composition without exploring interactions between microbes through co-occurrence network analysis, which can reveal mutualistic or competitive relationships within communities. Co-occurrence networks enable the identification of functional guilds, groups of microbes that collaborate to perform specific ecological functions, such as the degradation of complex compounds or sulfur oxidation.15,16 This approach is particularly relevant for the geothermal environment of Lake Linow, where microbial interactions are thought to be key to community resilience to environmental stress. In this study, we employed a metagenomic approach combined with co-occurrence network analysis to decipher the bacterial profile of hot mud from the Lake Linow geothermal area.
Research location
Hot mud samples from the Lake Linow exploration area in Tomohon. Samples were taken from three locations (Figure 1). Metagenomic analysis was conducted at the 1st BASE Laboratory, Axil Scientific Pte. Ltd., Singapore. Total genomic DNA isolation was conducted at the Molecular Biology Laboratory, Faculty of Mathematics and Natural Sciences, Manado State University.

Figure 1. Research Location and Sampling of Hot Mud in the Geothermal Area of Lake Linow, Tomohon, North Sulawesi, Indonesia
Research Procedures
Hot Mud Extraction
Sampling was conducted in the geothermal area of Lake Linow (1.270°N 124.827°E), Tomohon City, North Sulawesi, Indonesia. Prior to sampling, in situ measurements of physicochemical parameters were conducted, including temperature, pH, electrical conductivity, and total dissolved solids. In addition, major dissolved minerals and metals were quantified to characterize the geochemical conditions of the sampling sites (Table 1). The parameters observed were temperature and pH. The temperature parameter was measured using a thermometer inserted into the mud and left for 1 minute. The pH parameter was carried out using a pH meter paper dipped in the water surface. Mud samples were taken from the boiling mud area, at a depth of 50 cm from the mud surface and then preserved in a thermos. The mud samples were then taken to the Molecular Biology Laboratory, Faculty of Mathematics, Natural Sciences and Earth Sciences, Manado State University for total bacterial genomic isolation.17,18
Total DNA genomics extraction
Total genomic DNA from the hot mud was extracted using the Power Soil DNA Isolation Kit (MO BIO Laboratories, Carlsbad, CA, USA). The procedure followed the manufacturer’s standard protocol. The extraction results were then quantitatively analyzed using a Nano Drop and PicoGreen Assay to dsDNA 2000 spectrophotometer (Thermo Scientific, Wilmington, DE, USA). The concentration of extracted genomic DNA was quantified using the Quant-iT™ PicoGreen® dsDNA assay (Thermo Fisher Scientific), following the manufacturer’s instructions. Briefly, PicoGreen dye, which selectively binds to double-stranded DNA, was mixed with diluted DNA samples, and fluorescence intensity was measured using a fluorescence microplate reader. DNA concentrations were calculated based on a standard curve generated from known dsDNA standards.
Genomic DNA concentration was primarily quantified using the Quant-iT™ PicoGreen® dsDNA assay, which is less affected by protein, RNA, or reagent contaminants. Nanodrop spectrophotometry was used only as a preliminary quality control tool to assess absorbance ratios (Figure 2).
Figure 2. 16S rRNA gene amplification using the 2nd stage PCR method
PCR reaction
The initial PCR reaction was prepared in 50 µL using Q5 Hot Start High Fidelity PCR 2X Master mix according to the standard two-step protocol recommended by the manufacturer. The reaction mixture consisted of 25 µL Q5 High-fidelity 2x master mix, 2.5 µL each of 10 µM forward and reverse primers, 5 µL DNA template and 15 µL Nuclease-free water. The reaction started with an initial denaturation at 98 °C for 30 seconds followed by 35 cycles: 98 °C for 10 seconds, 68 °C for 1 minute, a final extension at 68 °C for 2 minutes and, finally, a hold at 4. The quality and quantity of PCR products were measured by TapeStation 4200 by Agilent Technologies PicoGreen and Nanodrop. All successful PCR products were then purified using AMPure XP beads according to the Illumina metagenomic sequencing preparation protocol (Table 2).
A second-stage PCR reaction involving the addition of dual indices (Nextera XT i7 index and Nextera XT i5 index) was prepared in 50 µL batches. The reaction mixture included 5 µL of the first step PCR product, 5 µL each of Nextera XT i7 and Nextera XT i5 index, 25 µL of high-fidelity 2x master mix Q5, and 10 µL of nuclease-free water (Table 2 and Table 3). The reaction began with an initial denaturation at 95 °C for 3 minutes, followed by 8 cycles of 95 °C for 30 seconds, 55 °C for 30 seconds, and 72 °C for 30 seconds, followed by a final extension at 72 °C for 5 minutes before holding at 40 °C. The quality and quantity of the final product were assessed using Agilent Technologies’ TapeStation 4200, PicoGreen, and qPCR. These samples were normalized to 4 nM and then pooled in equal volumes at 5 µL to yield a final 4 nM batch. The final pooled library was diluted and then denatured to a loading concentration of 4 pM with a 10% spiked-in PhiX control according to the protocol from Illumina and paired-end generated in 2×300 bp format from MiSeq (Figure 3).
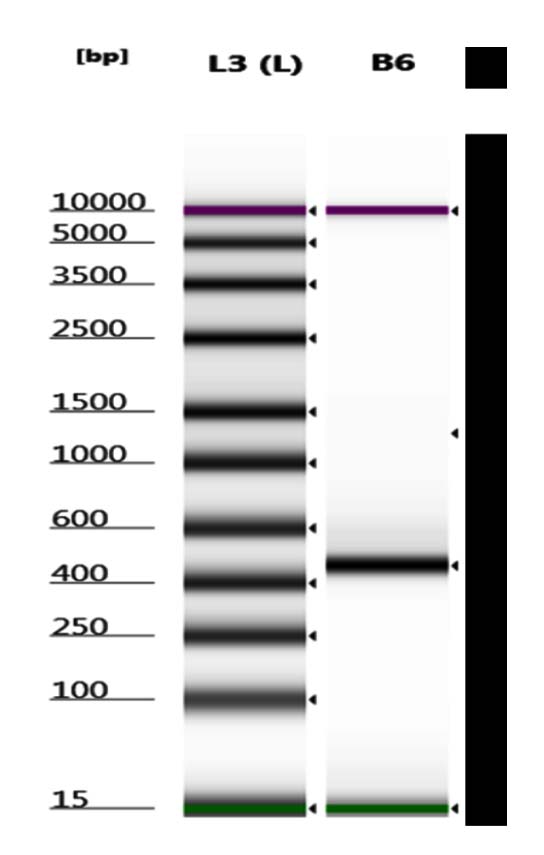
Figure 3. Sample Electrogram of PCR visualization of 16S rRNA gene in V3 and V4 areas
Metagenomic sequencing
Metagenomic sequencing was performed at 1st BASE Axil Scientific Pte. Ltd., (20-0200922-D) 41 Science Park Road, #04-08, The Gemini, Singapore Science Park II, Singapore. Amplicon Generation: Librarian barcoded V3 amplification was prepared using a two-step Illumina PCR protocol. The first stage of PCR amplification included the generation of PCR products targeting the V3 and V4 regions, followed by a second stage of PCR amplification performed with a sample-specific Illumina Nextera XT primer. A universal primer set targeting the V3 and V4 regions, with Illumina overhang adapters added at the 5′ and 3′ ends, was used.
The V3–V4 hypervariable regions of the 16S rRNA gene were amplified using universal primers with Illumina adapter overhang sequences. The forward primer sequence (341F) was 5′-TCGTCGGCAGC GTCAGATGTGTAT AAGAGACAGCCTACGGGNGGCWGCAG-3′ and the reverse primer sequence (805R) was 5′-GTCTCGTGGGCTCGGAGATGTGTATA AGAGACAGGACTACGVGGGTATCTAATCC-3′.
Bioinformatics analysis
Adapter sequences were processed using BBmap and merged using BBMerge from the packaged BBTools suite. Chimeras were then filtered using a search against the Greengenes database. All QC reads were clustered into Operational Taxonomic Units (OTUs) using UCLUST (de novo) at a similarity of 0.97. Representative sequences were extracted from each OTU for taxonomic assignment using UCLUST against the Greengenes database (release gg_13_8_99). Before taxonomic and diversity analyses, singletons and doubletons were removed, and samples were rarefied to the lowest number of reads among all samples.19 An internal reference normalization control (RN), provided by the sequencing facility, was included to monitor library preparation and sequencing performance and was not derived from environmental samples.
Extraction and purification of genomic DNA from hot mud
Total genomic DNA extraction was successfully performed using hot mud samples from the Lake Linow geothermal area in Tomohon. The genomic DNA concentration obtained was 20.4 ng/µl as measured by PicoGreen. Meanwhile, the genomic DNA concentration measured by Nanodrop was 23.21 ng/µl. DNA quantity suitable for metagenomic sequencing was confirmed using PicoGreen assay and successful PCR amplification of the 16S rRNA gene (Table 1).
Table (1):
Physicochemical characteristics of hot mud samples from Lake Linow at different sampling stations
| Parameter | Station I | Station II | Station III |
|---|---|---|---|
| In situ conditions (field measurement) | |||
| Temperature (°C) | 90 | 110 | 90 |
| Color | Black-grey | Black | Black-grey |
| Texture | Semi-solid | Semi-solid | Semi-solid |
| Salinity | 0.3 | 1.4 | 1.5 |
| Laboratory conditions (after transport) | |||
| Temperature (°C) | 37.3 | 36.0 | 37.3 |
| pH | 7.08 | 7.10 | 8.35 |
| Dissolved oxygen (mg L-1) | 7.16 | 7.40 | 7.69 |
| Color | Black | Brown | Black |
| Texture | Semi-solid | Semi-solid | Semi-solid |
Successful amplification of the 16S rRNA gene in the V3 and V4 regions was demonstrated by electrophoresis, which showed a stable band in well B6 (Figure 3). Visualization of the 16S rRNA gene amplicon determined the need for metagenomic PCR.
Amplicon library quality of PCR products
The results of 16S rRNA gene amplification of the V3 and V4 regions measured with PicoGreen showed a concentration of 22.4 ng/µl, while measurements with Nanodrop showed 24.44 ng/µl (Table 2). Based on metagenomic sequencing requirements, metagenomic sequencing can be continued (Table 3). Quality assessment of 16S rRNA gene libraries prepared for Illumina MiSeq sequencing. Library size and concentration were evaluated using TapeStation 4200 and PicoGreen assay. Libraries meeting Illumina sequencing requirements were classified as Pass. RN denotes an internal reference normalization control provided by the sequencing facility and is not a biological sample.
Table (2):
Extraction and purification of total genomic DNA from Lake Linow Hot Mud
No. |
Sample |
PicoGreen (ng/ul) |
Nanodrop (ng/ul) |
A260/A280 |
A260/A230 |
Description |
|---|---|---|---|---|---|---|
1 |
A2 |
20.4 |
23.21 |
1.54 |
0.25 |
Metagenomic sequencing continued |
Table (3):
Quality of PCR product amplicon library
| Sample | Concentration (ng/µL) | Pass/Fail | |
|---|---|---|---|
| PicoGreen | Nanodrop | ||
| RN | 75 | 550.18 | Pass |
| A2 | 22.4 | 24.44 | Pass |
16S rRNA library construction
The libraries were prepared using the Illumina 16S metagenomics library prep kit, and their quality and quantity were determined using the Agilent Tapestation 4200 and PicoGreen. The 16S rRNA library size was 607 bp with a concentration of 24.8 ng/nL, and was declared suitable for metagenomic sequencing. Library size (bp) was determined using Agilent TapeStation 4200 and represents the average fragment length of the 16S rRNA V3-V4 amplicon libraries.
Metagenomic sequencing and cluster generation
Metagenomic sequencing was performed in four reads. The total number of nucleotides read in metagenomic sequencing was 37,498,026. Cluster generation and a summary of the metagenomic sequencing implementation of mud from the geothermal area of Lake Linow, Tomohon are shown in Table 4 and Table 5.
Table (4):
Construction of 16S rRNA Library
Sample |
Library Size |
PicoGreen (ng/Ul) |
Molarity (Nm) Qpcr Method |
Pass/Fail |
|---|---|---|---|---|
A2 |
607 |
24.8 |
89.8 |
Pass |
Table (5):
Metagenomic sequencing and cluster generation
| Run summary for Sample A2 | ||||||
|---|---|---|---|---|---|---|
| Level | Yield Total (G) | Projected Total Yield (G) | Aligned (%) | Error Rate (%) | Intensity Cycle 1 | %>=Q30 |
| Read 1 | 5.02 | 5.02 | 6.46 | 2.76 | 69 | 76.65 |
| 53 Read 2 (1) | 0.12 | 0.12 | 0 | 0 | 370 | 85.9 |
| Read 3 (1) | 0.12 | 0.12 | 0 | 0 | 459 | 88.98 |
| Read 4 | 5.02 | 5.02 | 6.28 | 3.28 | 70 | 66.99 |
| Non-indexed Total | 10.05 | 10.05 | 6.37 | 3.02 | 70 | 71.82 |
| Total | 10.28 | 10.28 | 6.37 | 3.02 | 242 | 72.18 |
| Total Reads | Passing Filtered (PF) Reads | Total % Reads Identified (Pf) | ||||
| 37.498.026 | 33.483.944 | 86.90 | ||||
| Sample ID | Sample Name | % Reads Identified (PF) | ||||
| 10461-1 | A2 | 0.97 | ||||
Metagenomic bacterial grouping
Based on metagenomic analysis using the 16S rRNA gene, 15 families were found, with the largest number of species belonging to the Proteobacteria family (Figure 4).
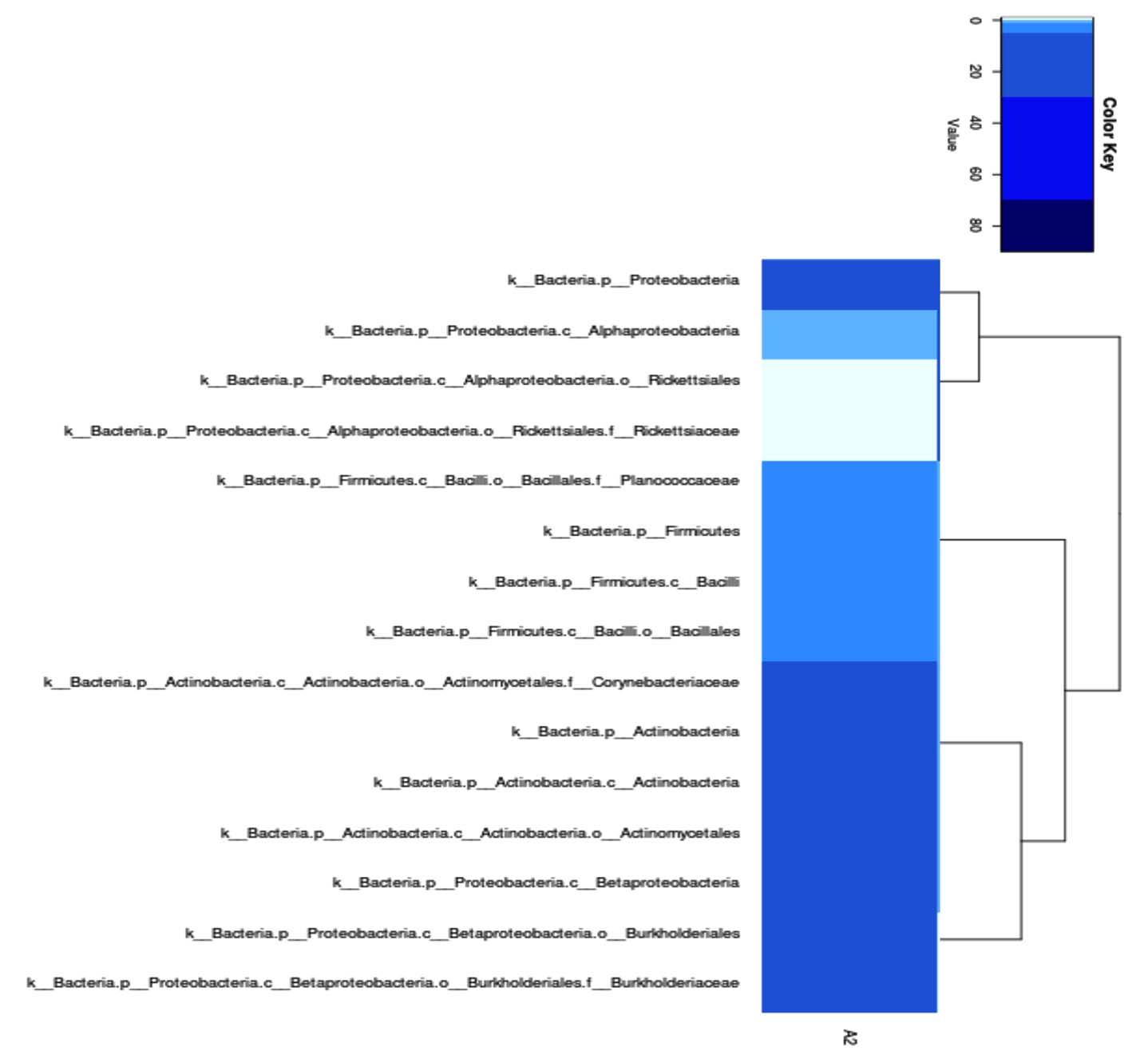
Figure 4. Family level phylogeny of metagenomic sequencing results of bacteria from hot mud
Metagenomic analysis at the genus level yielded 19 bacterial genera. The genera with the largest number of species were Proteobacteria, Alphaproteobacteria, Burkholderia, Actinobacteria, Corynebacterium, and Planocococaceae (Figure 5).
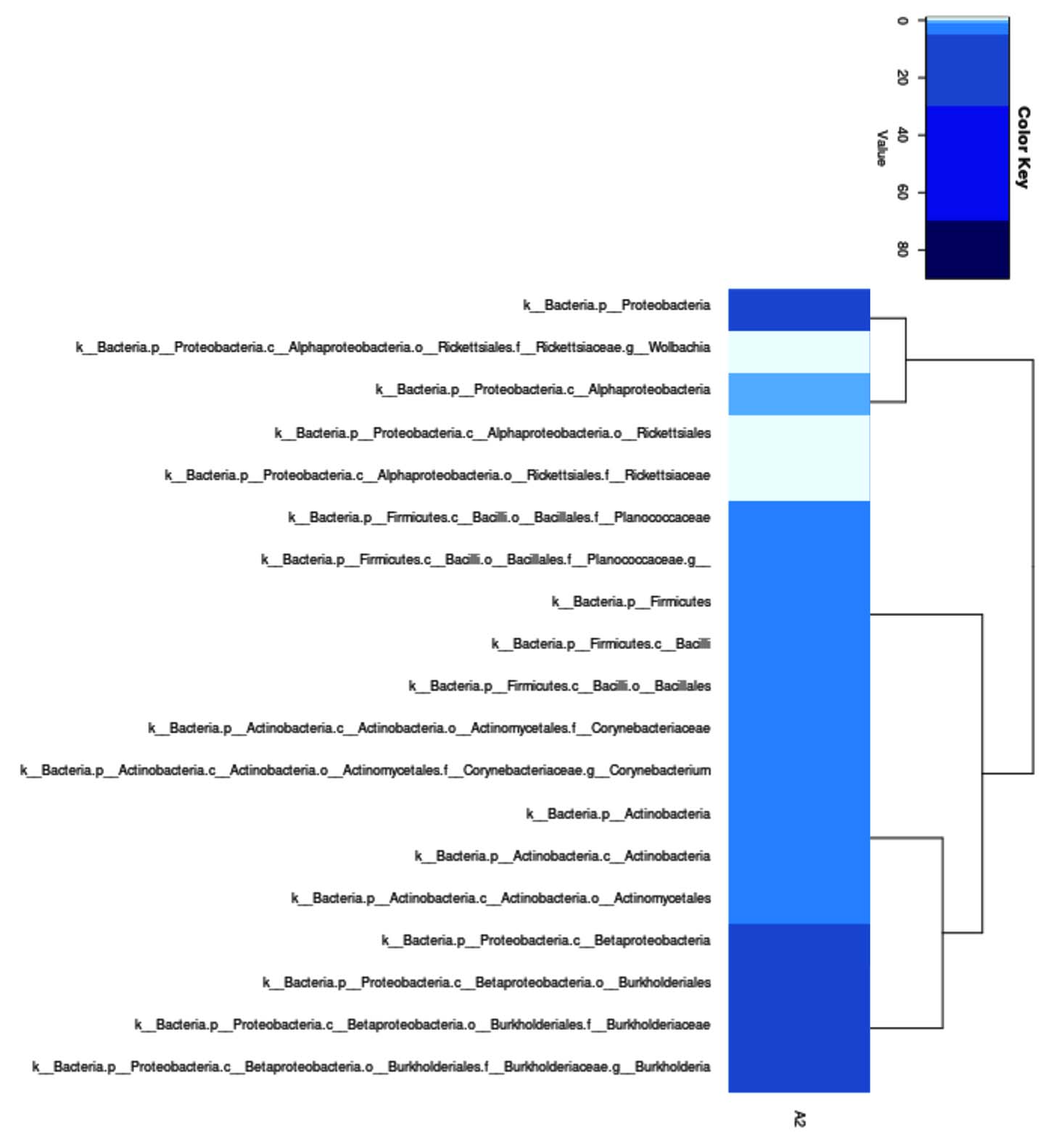
Figure 5. Genus level phylogeny of metagenomic sequencing results of bacteria from hot mud
Family and genus level phylogeny from metagenomic results
The phylogenetic tree resulting from metagenomic analysis is shown at the family and genus levels. Both the genus and family levels show two monophyletic groups. Based on abundance, Corynebacterium (Family) and Corynebacterium (Genus) show the highest abundance. The Caulobacteraceae and Isosphaeraceae families have not yet been identified as genera based on gene bank data. The family- and genus-level phylogenetic trees (Figure 6) highlight the dominance of Burkholderiaceae and Burkholderia, as reflected by both clustering patterns and symbol size.
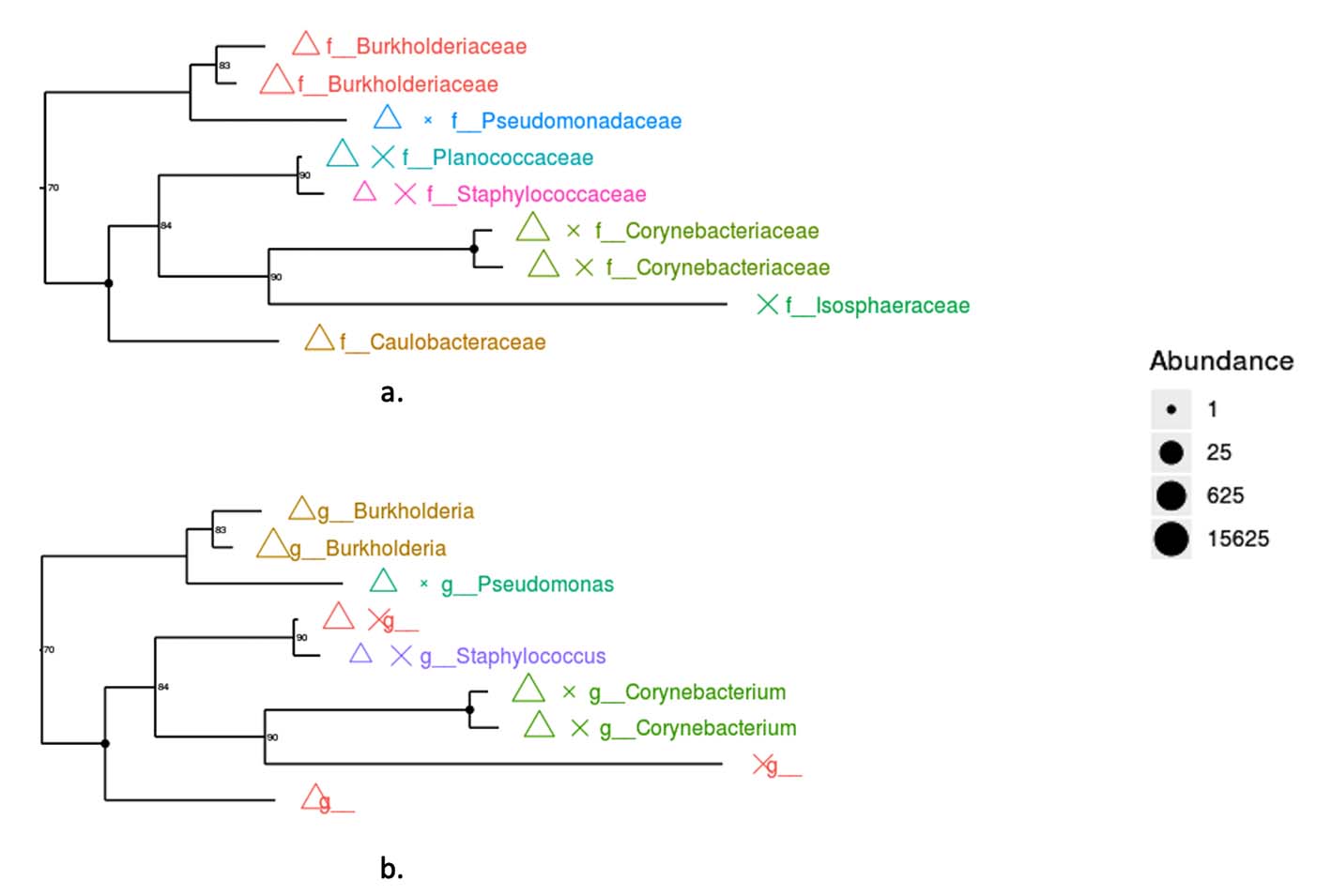
Figure 6. Phylogenetic tree based on 16S rRNA gene (a). Family level (b). Genus level. Colored symbols represent different bacterial families (a) and genera (b), while symbol size indicates relative abundance based on read counts. Triangles denote dominant taxa, whereas crosses represent less abundant taxa. Bootstrap values (>70%) are shown at internal nodes
The co-occurrence network of the 100 most abundant OTUs (Operational Taxonomic Units) in the metagenomic analysis of hot mud from the geothermal area of Lake Linow is shown in Figure 7. Each node in the network represents one OTU (a group of microbes based on the similarity of certain 16S rRNA gene sequences). The lines between the dots (edges) indicate the co-occurrence relationship between these OTUs. This network shows how various microbes associate with each other in a particular environment. Connecting lines can indicate: Mutualistic/symbiotic relationships, Overlapping ecological niches and Metabolic dependencies. The color distribution indicates which phylum is dominant in the metagenomic analysis of hot mud, namely Proteobacteria. Groups of closely connected nodes may indicate functional guilds or groups of closely interacting microbes.
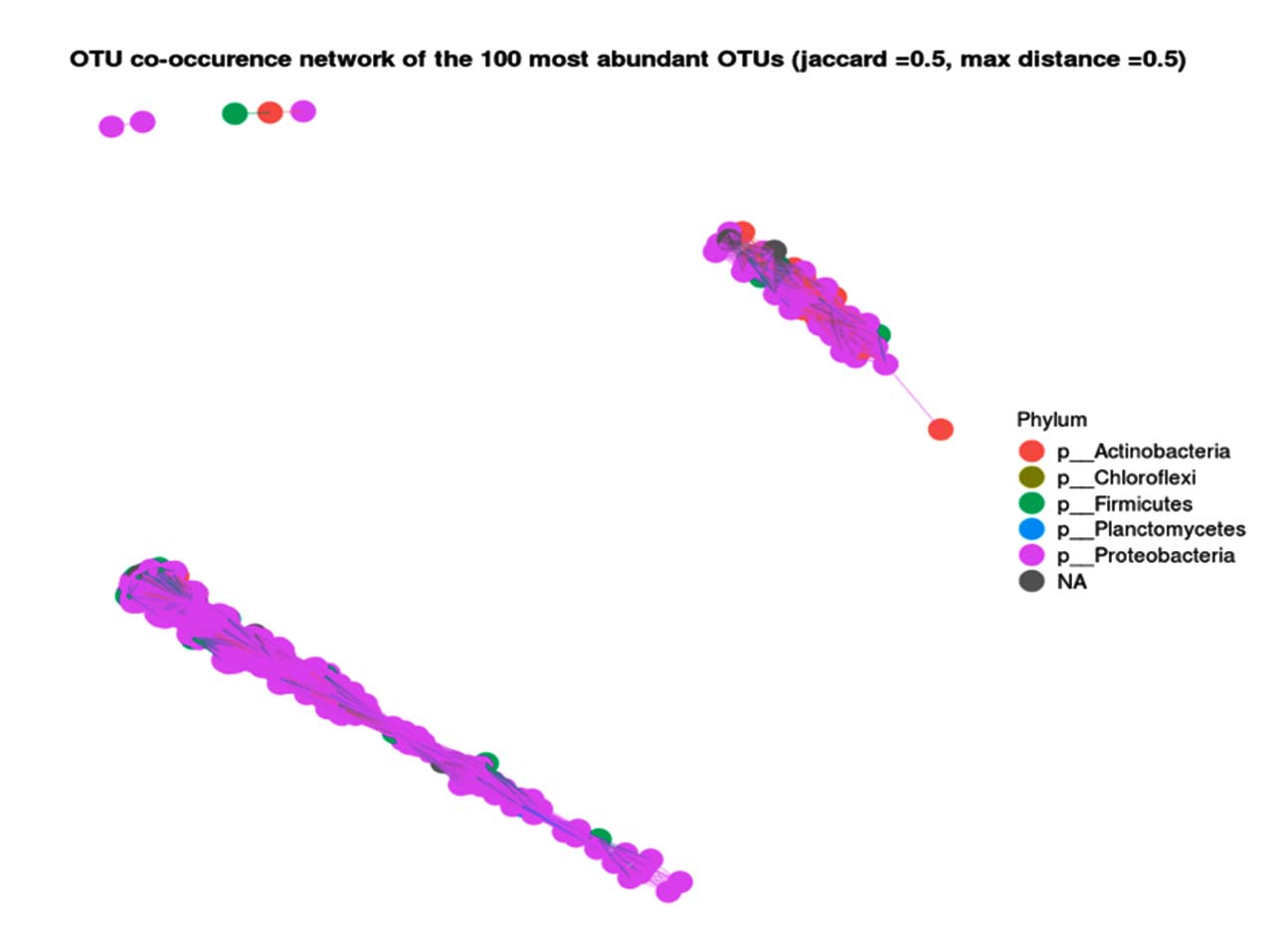
Figure 7. OTU co-occurrence network results from metagenomic analysis of hot mud
A hierarchy-based interactive visualization was used to display the microbial community composition from metagenomic analysis of hot mud from the Linow Lake Geothermal Area. The area or color in each segment indicates the relative proportion of a taxon in the sample. The genus Burkholderia (65%) dominated the hot mud metagenomic results, although approximately 0.05% of the bacteria were not yet recorded in the NCBI gene bank (Figure 8).
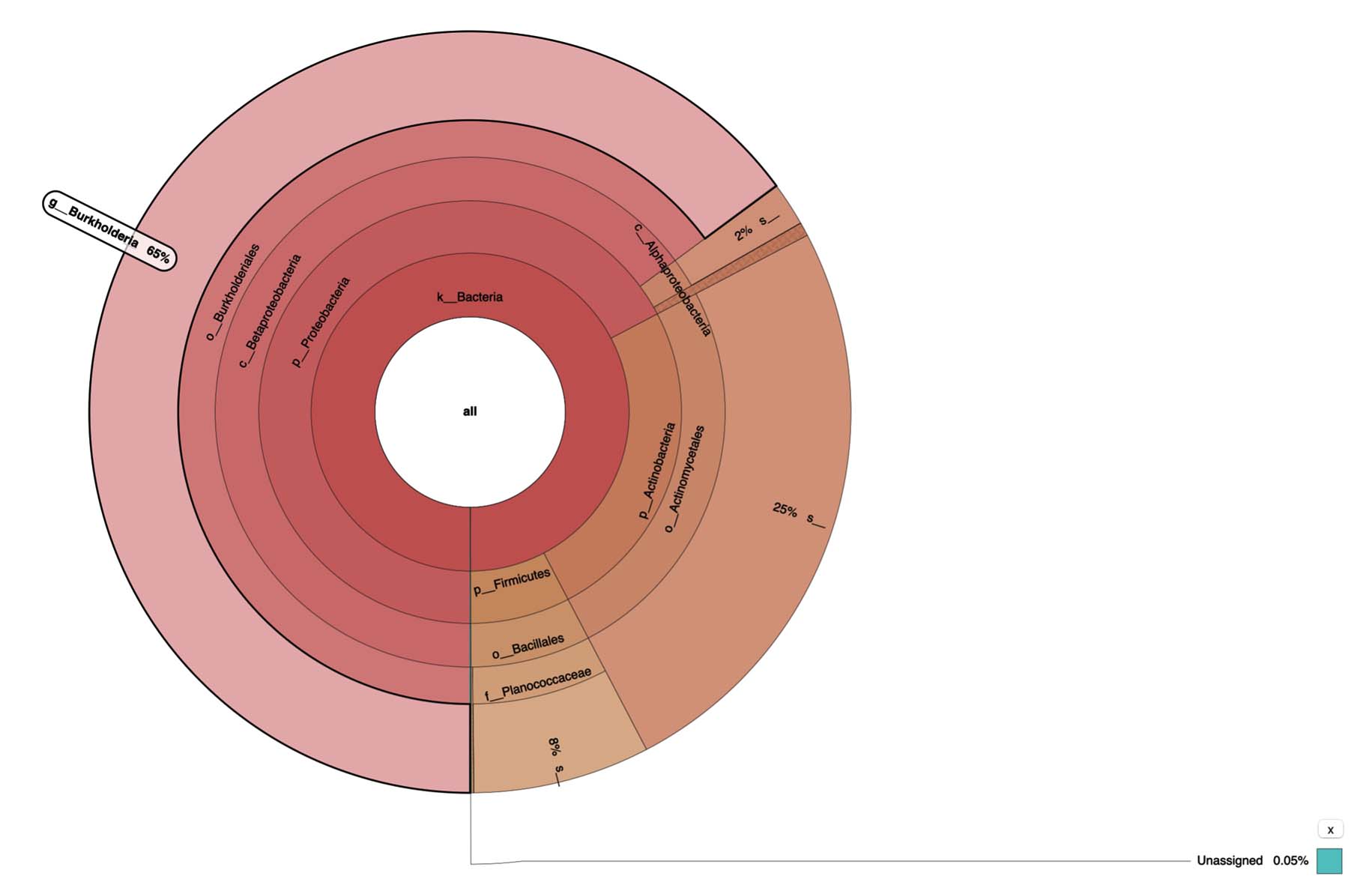
Figure 8. Krona results of metagenomic analysis of Hot Mud from Lake Linow
Bacterial composition at family level
The family level composition of metagenomic analysis results based on the 16S rRNA gene, hot mud samples found 0.1% of the Unassigned bacterial population, which means it has not been recorded or identified in the world’s bacterial taxonomy system. The bacterial database based on the 16S rRNA gene has been stored in the NCBI gene bank. A total of 65% belongs to the Burkholderiaceae family; 24.6% belongs to the Corynebacteriacea family; 7.6% belongs to the Planococcaceae family; 1.9% belongs to the Caulobacteraceae family; 0.5% belongs to the Pseudomanadaceae family and 0.1% belongs to the Methanobacteriaceae family. Thus, the Burkholderiaceae family is the dominant bacterial family in hot mud samples from the geothermal area of Lake Linow (Figure 9).
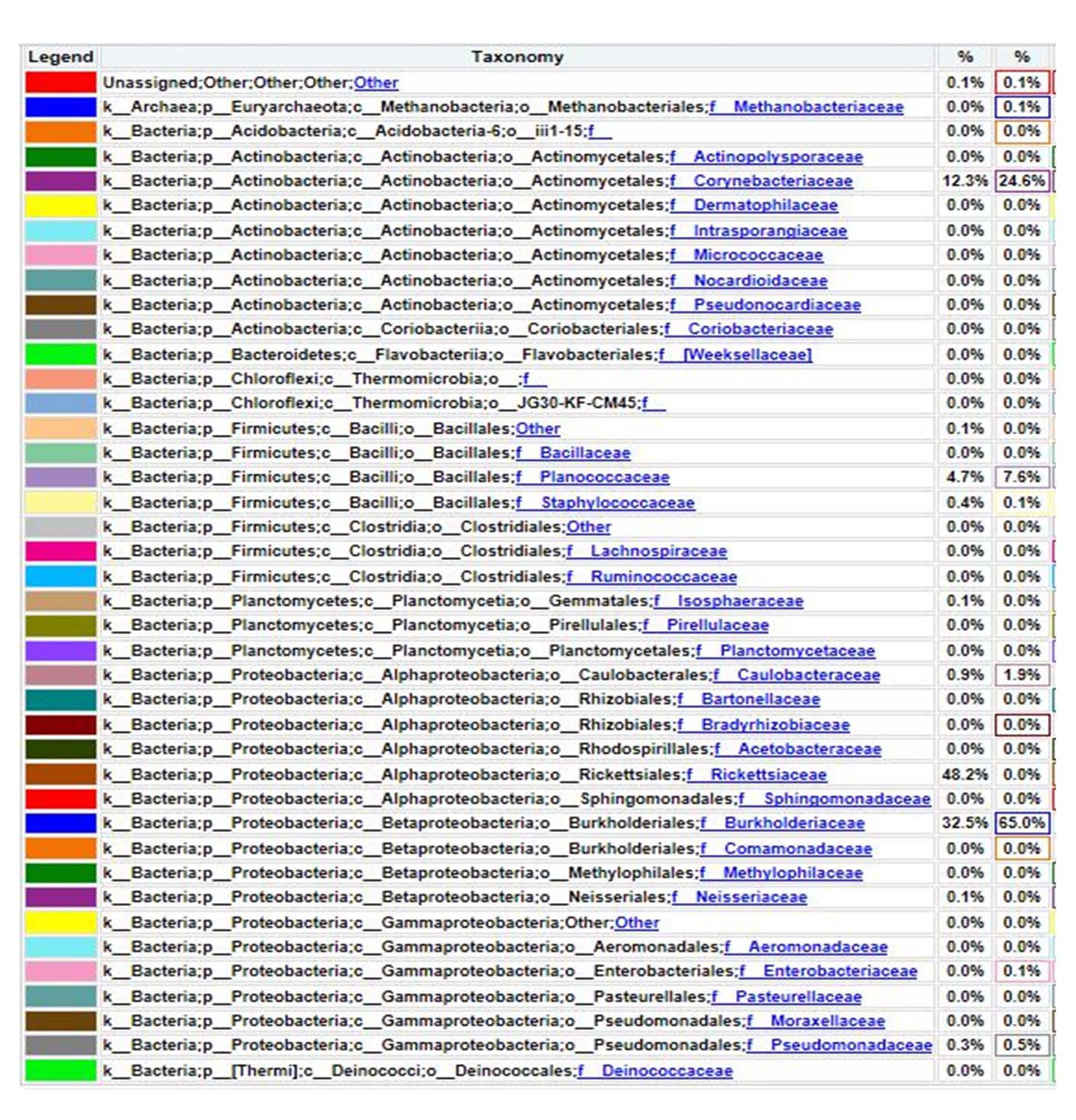
Figure 9. Bacterial composition at family level
Bacterial composition at genus level
The genus level composition of metagenomic analysis results based on the 16S rRNA gene, found 0.1% of the Unassigned bacterial population, which means it has not been recorded or identified in the world’s bacterial taxonomy system. The bacterial database based on the 16S rRNA gene has been stored in the NCBI gene bank. A total of 24.6% are included in the Corynebacteriaceae genus; 7.6% are included in the Planococcaceae family but there is no precise data on the genus group; 1.9% are included in the Caulobacteraceae family and there is no precise data on the genus group; 65% are included in the Burkholderia genus; 0.1% are included in the Methanobacterium genus. Thus, the Burkholderia family is the dominant bacterial genus in hot mud samples from the geothermal area of Lake Linow (Figure 10).
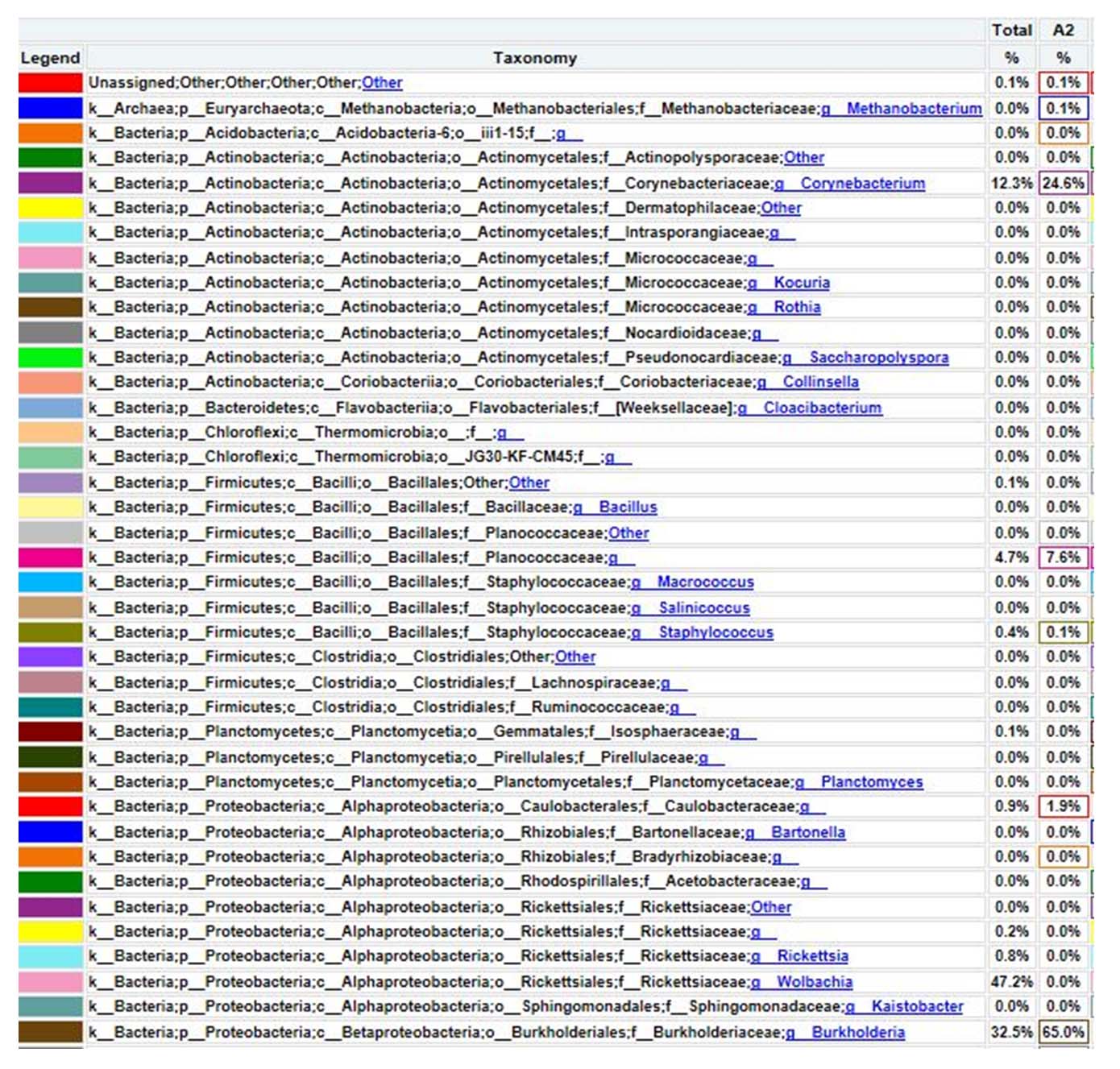
Figure 10. Bacterial composition at genus level
Based on metagenomic analysis using the 16S rRNA gene, 15 bacterial families were found in hot mud samples from the geothermal area of Lake Linow, with the Proteobacteria family as the most dominant group. The dominance of Proteobacteria indicates high adaptation to extreme environments.20,21 These Proteobacteria are known to have a wide metabolic diversity, including chemolithotrophy and the ability to survive in hot conditions and high sulfur content.22 Several genera in Proteobacteria, such as Thiobacillus and Acidithiobacillus, are often found in geothermal environments due to their ability to oxidize sulfur and iron compounds,23 which may play a role in the biogeochemical cycle in Lake Linow. Furthermore, the presence of other families such as Firmicutes and Chloroflexi indicates the presence of thermophilic and anaerobic microbes that play a role in organic matter decomposition and the carbon cycle in these hot environments.24
Thermophilic bacteria, including some Proteobacteria, have evolved mechanisms to thrive at high temperatures.25 These adaptations involve maintaining membrane stability, protecting nucleic acids, and ensuring protein functionality.26,27 Thermophilic proteins exhibit enhanced stability compared to mesophilic counterparts, achieved through specific amino acid compositions that minimize biosynthesis costs and optimize chaperone activity.28 Some thermophiles, such as those in the genus Thermus, have been metabolically engineered to grow anaerobically, expanding their potential applications.29 Thermophilic cyanobacteria serve as models for studying heat tolerance in photosynthetic organisms, offering insights into molecular mechanisms of thermoadaptation.29 The ability to withstand high temperatures often correlates with metal resistance, making thermophiles valuable for various industrial processes, including bioremediation of heavy metal-contaminated environments.30
These results also reveal the potential for complex microbial interactions in the hot mud ecosystem of the Linow Lake geothermal area. The dominance of Proteobacteria may correlate with nutrient availability and physicochemical conditions, such as low pH and high temperature, which are common in geothermal areas. The presence of families such as Actinobacteria and Planctomycetes suggests a role in the degradation of complex organic compounds and the nitrogen cycle. Further analysis using functional prediction or complete metagenomic sequencing approaches could reveal more details about the active metabolic pathways, as well as their biotechnological potential, such as thermostable enzymes or bioremediation applications. These findings strengthen our understanding of microbial diversity in Indonesia’s geothermal ecosystems and their contribution to biogeochemical processes.
Metagenomic analysis of Lake Linow hot mud samples revealed 19 bacterial genera, dominated by the Proteobacteria (class Alphaproteobacteria) and Burkholderia, consistent with findings in other geothermal environments.31,32 The genus Burkholderia is widely recognized as a group of bacteria that adapts to a wide range of environments, including extreme conditions such as high temperatures.33 The genus Burkholderia is known for its adaptability to diverse environments, including extreme conditions like high temperatures.34 Burkholderia species can survive in a variety of habitats, from soil and water to plant tissues. Although most Burkholderia species are mesophilic, some members exhibit thermophilic or thermotolerant properties.
The abundant presence of Alpha-proteobacteria suggests an important role in the sulfur and carbon cycles, supported by their diverse metabolic capabilities, including chemosynthesis and adaptation to thermophilic conditions.35 The genus Burkholderia, known for its ability to degrade complex organic compounds and resistance to heavy metals, indicates potential bioremediation in these extreme environments. One example of a member of the genus Burkholderia is Burkholderia thermophila. Burkholderia thermophila grows optimally at temperatures of 50-55 °C; this bacterium possesses genes encoding thermally stable chaperone proteins (e.g., Hsp60).36 In addition, Actinobacteria and Corynebacterium, often associated with the production of bioactive compounds and adaptation to acidic environments,37 as well as Planococcaceae, a group of heat-resistant Gram-positive bacteria that play a role in the decomposition of organic material.
The identified genus composition reflects microbial adaptation to the extreme physicochemical conditions of Lake Linow, characterized by high temperatures
(68-95 °C), acidic pH (2.2-3.4), and elevated sulfur-related minerals (Table 1). The dominance of Proteobacteria and Alphaproteobacteria is consistent with studies in Yellowstone (USA) and Iceland hot springs,38 indicating the functional conservation of geothermal microbes globally. The presence of Actinobacteria and Planococcaceae strengthens the hypothesis that this environment harbors genetic diversity for thermostable enzymes and antibacterial compounds.39,40 The discovery of the genus Corynebacterium, typically associated with soil and the rhizosphere, suggests possible gene flow from the surrounding environment.41 Further studies through whole-genome sequencing and metabolomic analysis may reveal specific adaptation mechanisms and potential biotechnological applications, such as biomining or industrial enzyme production. These results provide a basis for further exploration of geothermal microbial diversity in Indonesia, which remains underexplored compared to other locations worldwide.
16S rRNA gene-based metagenomic analysis of Lake Linow’s thermal mud revealed a unique microbial composition, dominated by Burkholderiaceae (65%), followed by Corynebacteriaceae (24.6%), Planococcaceae (7.6%), Caulobacteraceae (1.9%), Pseudomonadaceae (0.5%), and Methanobacteriaceae (0.1%), with 0.1% of the population unclassified (Unassigned). The dominance of Burkholderiaceae is in line with findings in other acidic geothermal environments, where this group plays a role in sulfur cycling and adaptation to heavy metals.42,43 The high abundance of Corynebacteriaceae, known to produce bioactive compounds, suggests potential bioprospecting for pharmaceutical applications.44 The presence of 0.1% “Unassigned” sequences indicates the possibility of new taxa that have not been recorded in the reference database, emphasizing the importance of exploring microbial diversity in Indonesia’s geothermal ecosystems which are still underexplored.
Co-occurrence network analysis of the 100 most abundant OTUs in Lake Linow’s thermal mudflats revealed a complex microbial community structure, dominated by the phylum Proteobacteria (indicated by the node color distribution). The observed relationship patterns, including highly clustered node groups, suggest the presence of ecologically interacting microbial functional guilds, likely related to sulfur and carbon cycling in geothermal environments. This finding aligns with research from Yellowstone Hot Springs (USA) that reported that Proteobacteria form a strong co-occurrence network module, driven by a metabolic dependence on sulfur compounds and high temperatures.45 The mutualistic relationships among OTUs within this group may reflect metabolic syntrophy, as observed in hydrogen sulfide-producing microbial consortia.46 The presence of overlapping ecological niches between Proteobacteria OTUs and other phyla such as Actinobacteria and Chloroflexi (different colored nodes) indicates resource partitioning or collaboration in the degradation of complex organic matter.
Metagenomic analysis of Lake Linow’s hot mud revealed a unique microbial community dominated by the phylum Proteobacteria, particularly the families Burkholderiaceae (65%) and Corynebacteriaceae (24.6%), with important roles in sulfur cycling, adaptation to extreme conditions (high temperature, acidity, and heavy metals), and potential for bioactive compound production. The co-occurrence network revealed complex interactions among OTUs, including mutualistic relationships, ecological niche partitioning, and metabolic interdependence, forming functional guilds related to organic matter degradation and inorganic compound oxidation. The presence of 0.1% “unassigned” sequences indicates the possibility of new, unidentified taxa, emphasizing the importance of further exploration using whole-genome sequencing approaches. These findings not only enhance our understanding of Indonesia’s underexplored geothermal microbial diversity but also highlight its biotechnological potential, such as thermostable enzymes from the Planococcaceae and Actinobacteria groups, heavy metal bioremediation by Burkholderiaceae, and bioactive compounds from Corynebacteriaceae. Further studies with metatranscriptomic and metabolomic analyses are needed to uncover the specific interaction mechanisms and practical applications of these microbial communities.
ACKNOWLEDGMENTS
The authors would like to express their deepest gratitude to the Rector of Universitas Negeri Manado and the Institute of Research and Community Service (LPPM) of Universitas Negeri Manado for their financial support that made this research possible. The authors also wish to thank the Biology Laboratory for offering the essential facilities and technical support during the course of this study.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
AUTHORS’ CONTRIBUTION
HMS and MYS conceptualized the study, applied methodology and investigated the study. MYS performed data and formal analysis and collected the resources. HMS and MYS wrote the original draft. MYS and HMS wrote, reviewed, and revised the manuscript. HMS performed supervision. All authors read and approved the final manuscript for publication.
FUNDING
This research was funded by LPPM State University of Manado in 2024, with the main contract number 192/ES/PG.02.00.PL/2025, through the Fundamental Research scheme.
DATA AVAILABILITY
All datasets generated or analyzed during this study are included in the manuscript.
ETHICS STATEMENT
Not Applicable.
- Ali JR, Heaney LR. Wallace’s line, Wallacea, and associated divides and areas: history of a tortuous tangle of ideas and labels. Biol Rev. 2021;96(3):922-942.
Crossref - Heads M, Grehan JR, Nielsen J, Patrick B. Biogeographic-tectonic calibration of 14 nodes in a butterfly timetree. Cladistics. 2023;39(4):293-336.
Crossref - Jolie E, Scott S, Faulds J, et al. Geological controls on geothermal resources for power generation. Nat Rev Earth Environ. 2021;2(5):324-339.
Crossref - Sousa J, Silverio SC, Costa AMA, Rodrigues LR. Metagenomic Approaches as a Tool to Unravel Promising Biocatalysts from Natural Resources: Soil and Water. Catalysts. 2022;12(4):385.
Crossref - Hadland N, Hamilton CW, Duhamel S. Young volcanic terrains are windows into early microbial colonization. Commun Earth Environ. 2024;5(1):114.
Crossref - Harindintwali JD, Xiang L, Wang F, et al. Syntrophy of bacteria and archaea in the anaerobic catabolism of hydrocarbon contaminants. Crit Rev Environ Sci Technol. 2022;53(13):1331-1357.
Crossref - Sandalayuk E, Natsir H, Arfah RA, Wahab AW, Taba P, Rasyid H. Exploring the diversity and bioremediation potential of thermohalophilic bacteria from Wawolesea geothermal spring, southeast Sulawesi, Indonesia. Ecological Engineering & Environmental Technology (EEET). 2025;26(7):179-185.
Crossref - Yan B, Li X, Qiao N, Da Z, Xu J, Jiang C, Ba S. The co-occurrence patterns and assembly mechanisms of microeukaryotic communities in geothermal ecosystems of the Qinghai-Tibet Plateau. Front Microbiol. 2025;16:1513944.
Crossref - Suddin S, Mokosuli YS, Marcelina W, Orbanus N, Ardi K. Molecular barcoding based 16S rRNA gene of Thermophilic bacteria from vulcanic sites, Linow Lake, Tomohon. Materials Science Forum. 2019;967:83-92.
Crossref - Chaudhary S, Sindhu SS, Dhanker R, Kumari A. Microbes-mediated sulphur cycling in soil: Impact on soil fertility, crop production and environmental sustainability. Microbiol Res. 2023;271:127340.
Crossref - Bazzicalupo AL, Erlandson S, Branine M, et al. Fungal community shift along steep environmental gradients from geothermal soils in Yellowstone National Park. Microb Ecol. 2022;84(1):33-43.
Crossref - Nordgard-Hansen E, Fjellsa IF, Medgyes T, et al. Differences in Direct Geothermal Energy Utilization for Heating and Cooling in Central and Northern European Countries. Energies. 2023;16(18):6465.
Crossref - Shomali BA, Danish-Daniel M. Review on bioprospecting of thermophilic enzymes from hot springs via Omics approaches. Aquaculture, Aquarium, Conservation & Legislation. 2024;17(5):2156-2174.
- Sadeepa D, Sirisena K, Manage PM. Diversity of microbial communities in hot springs of Sri Lanka as revealed by 16S rRNA gene high-throughput sequencing analysis. Gene. 2022;812:146103.
Crossref - Siles JA, Garcia-Sanchez, M, Gomez-Brandon M. Studying Microbial Communities through Co-Occurrence Network Analyses during Processes of Waste Treatment and in Organically Amended Soils: A Review. Microorganisms. 2021;9(6):1165.
Crossref - Ishimoto CK, Aono AH, Nagai JS, et al. Microbial co-occurrence network and its key microorganisms in soil with permanent application of composted tannery sludge. Sci Total Environ. 2021;789:147945.
Crossref - Babalola OO, Olowe OM, Ayangbenro AS. Shotgun metagenomics dataset of Striga hermonthica-infested maize (Zea mays L.) rhizospheric soil microbiome. Data in Brief. 2023;48:109132.
Crossref - Das S, Kumari A, Sherpa MT, Najar IN, Thakur N. Metavirome and its functional diversity analysis through microbiome study of the Sikkim Himalayan hot spring solfataric mud sediments. Curr Res Microb Sci. 2020;1:18-29.
Crossref - Xia Y, Li X, Wu Z, et al. Strategies and tools in illumina and nanopore integrated metagenomic analysis of microbiome data. IMeta. 2023;2(1):e72.
Crossref - Li YQ, Chai YH, Wang XS, et al. Bacterial community in saline farmland soil on the Tibetan plateau: responding to salinization while resisting extreme environments. BMC Microbiol. 2021;21(1):119.
Crossref - Gao L, Huang Y, Liu Y, et al. Bacterial Community Structure and Potential Microbial Coexistence Mechanism Associated with Three Halophytes Adapting to the Extremely Hypersaline Environment. Microorganisms. 2022;10(6):1124.
Crossref - Keller LM, Colman DR, Boyd ES. Simultaneous aerobic and anaerobic respiration in hot spring chemolithotrophic bacteria. Nat Commun. 2025;16(1):1063.
Crossref - Ogola HJO, Selvarajan R, Ncube S, Madikizela L. Thiocapsa, Lutimaribacter, and Delftia Are Major Bacterial Taxa Facilitating the Coupling of Sulfur Oxidation and Nutrient Recycling in the Sulfide-Rich Isinuka Spring in South Africa. Biology. 2025;14(5):503.
Crossref - Liu X, Zhong L, Yang R, et al. Modifying soil bacterial communities in saline mudflats with organic acids and substrates. Front Microbiol. 2024;15:1392441.
Crossref - Yaman BN. 16S amplicon sequencing and functional gene prediction of microbial community in Inner West Aegean, Turkey. Comput Biol Chem. 2025;115:108373.
Crossref - Karampatakis T, Tsergouli K, Behzadi P. Carbapenem-resistant Pseudomonas aeruginosa’s resistome: pan-genomic plasticity, the impact of transposable elements and jumping genes. Antibiotics. 2025;14(4):353.
Crossref - Susanto I, Jayanegara A, Ridwan R, Wiryawan IKG, Laconi EB. Metagenomic insights into microbial community dynamics of fermented Indigofera zollingeriana supplemented with probiotics and phytobiotics. Biodiversitas Journal of Biological Diversity. 2025;26(6).
Crossref - Jaiswal N, Jaiswal P. Thermostable a-Amylases and Laccases: Paving the Way for Sustainable Industrial Applications. Processes. 2024;12(7):1341.
Crossref - Aggarwal H, Chaudhary D, Tyagi J, et al. Thermophiles and Their Diverse Function in Agricultural and Biotechnological Applications. In: Ranjan A, Rajput VD, Chauhan A, Prazdnova EV, Minkina T, Zargar SM. (eds) Extremophiles for Sustainable Agriculture and Soil Health Improvement. Springer, Cham.
Crossref - Rasul F, You D, Jiang Y, Liu X, Daroch M. Thermophilic cyanobacteria-exciting, yet challenging biotechnological chassis. Appl Microbiol Biotechnol. 2024;108(1):270.
Crossref - Chia XK, Hadibarata T, Jusoh MNH, Tan IS, Foo HCY. Extremophilic Microbes for Environmental Bioremediation: A Review. Biointerface Res Appl Chem. 2024;14(5):103.
Crossref - Gallo A, Sposito F, Longo M, et al. Perturbations in Microbial Communities at Hydrothermal Vents of Panarea Island (Aeolian Islands, Italy). Biology. 2025;14(1):86.
Crossref - Ibrahim NH, Atef A, Abd El-Rahman MA, Sidkey NM. A review on thermo-alkalophilic bacterial lipase production and its applications. Int J Theor Appl Res. 2025;4(1).
Crossref - Gulati A, Thakur R, Vyas P, et al. Fostering climate-resilient agriculture with ACC-deaminase producing rhizobacterial biostimulants from the cold deserts of the Indian Himalayas. J Environ Manage. 2024;371:123075.
Crossref - Li J, Xiang S, Li Y, et al. Arcobacteraceae are ubiquitous mixotrophic bacteria playing important roles in carbon, nitrogen, and sulfur cycling in global oceans. Msystems. 2024;9(7):e00513-24.
Crossref - Dukat AM, Elcheninov AG, Klyukina AA, Novikov AA, Frolov EN. Thiobacter aerophilum sp. nov, a Thermophilic, Obligately Chemolithoautotrophic, Sulfur-Oxidizing Bacterium from a Hot Spring and Proposal of Thiobacteraceae fam. nov. Microorganisms. 2024;12(11):2252.
Crossref - Muhammad M, Rekadwad BN, Habib T, Dong L, Hozzein WN, Li WJ. Applications of Bioactive Compounds from Novel Microbial Taxa. In: Li WJ, Jiao Jy, Salam N, Rao MPN. (eds) Modern Taxonomy of Bacteria and Archaea. Springer, Singapore. 2024:195-208.
Crossref - Sriaporn C, Campbell KA, Van Kranendonk MJ, Handley KM. Bacterial and archaeal community distributions and cosmopolitanism across physicochemically diverse hot springs. ISME Communications. 2023;3(1):80.
Crossref - Farda B, Djebaili R, Vaccarelli I, Del Gallo M, Pellegrini M. Actinomycetes from Caves: An Overview of Their Diversity, Biotechnological Properties, and Insights for Their Use in Soil Environments. Microorganisms. 2022;10(2):453.
Crossref - Helmi NR. Exploring the diversity and antimicrobial potential of actinomycetes isolated from different environments in Saudi Arabia: a systematic review. Front Microbiol. 2025;16:1568899.
Crossref - Petrovic M, Janakiev T, Grbic ML, et al. Insights into endophytic and rhizospheric bacteria of five sugar beet hybrids in terms of their diversity, plant-growth promoting, and biocontrol properties. Microb Ecol. 2024;87(1):19.
Crossref - Mitrovic M, Kostesic E, Markovic T, et al. Microbial community composition and hydrochemistry of underexplored geothermal waters in Croatia. Syst Appl Microbiol. 2022;45(6):126359.
Crossref - Rowe L, Dowd SE, Davidson K, et al. Comparing microbial populations from diverse hydrothermal features in Yellowstone National Park: hot springs and mud volcanoes. Front Microbiol. 2024;15:1409664.
Crossref - Priscilla A, Achudhan AB, Saleena LM. Advances in Microbial Metagenomics for Bioprospecting in Nutraceuticals and Pharmaceuticals. Curr Pharmacol Rep. 2025;11(1):38.
Crossref - Peng X, Wang S, Wang M, et al. Metabolic interdependencies in thermophilic communities are revealed using co-occurrence and complementarity networks. Nat Commun. 2024;15(1):8166.
Crossref - Bae I, Rhee C, Shin J, Cho K, Triolo JM, Shin SG. Insights into high ammonia-resistant syntrophic microbiomes and metabolic pathways during continuous anaerobic digestion of cow manure. Bioresour Technol. 2025;422:132235.
Crossref
© The Author(s) 2026. Open Access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License which permits unrestricted use, sharing, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.