


Bacterial persisters are a tiny group of bacterial cells that manage to survive antibiotic exposure without becoming genetically resistant. These cells slip into a temporary resting state that protects them from drugs and lets the infection come back later. Their formation is influenced by many stress factors such as antibiotics, the host’s immune molecules like reactive oxygen and nitrogen species, a lack of nutrients, and the slow-growth conditions found in biofilms. Inside the cell, several survival systems work together including the stringent response, toxin-antitoxin activity, ribosome hibernation, protein and DNA repair mechanisms, and changes in energy balance to keep the bacteria dormant but alive. Studies using single-cell tools show that not all persisters behave the same; some wake up quickly when the stress is gone, while others take much longer. The return to activity mainly depends on the recovery of energy, protein quality, and translation processes, all of which are shaped by signals from the surrounding environment and host tissues. Understanding how these cells go to sleep and wake again has led to new treatment ideas such as forcing them to wake before applying antibiotics (“wake-and-kill”) or targeting them while they are still inactive (“kill-in-sleep”). Even with these advances, it remains difficult to clearly define persisters or to turn lab findings into reliable clinical treatments.
Bacterial Persisters, Phenotypic Antibiotic Tolerance, Stringent Response, Biofilm Persistence, Antimicrobial Resistance
Antibiotic persistence has emerged as a central theme in microbial pathogenesis and treatment failure, distinct from classical genetic resistance. Unlike resistant mutants, persisters are phenotypically tolerant subpopulations that transiently withstand otherwise lethal drug concentrations while remaining genetically susceptible. This survival is typically achieved through dormancy, metabolic slowdown, ribosome hibernation, or activation of stress-response pathways, enabling cells to survive prolonged therapy and subsequently reseed infection once antibiotic pressure subsides.1 The recognition of persisters has reframed how chronic and relapsing infections are understood, with direct implications for diseases caused by Staphylococcus aureus, Pseudomonas aeruginosa, Escherichia coli, and Mycobacterium tuberculosis.2
Recent advances have challenged the traditional view of persisters as uniformly dormant. Single-cell approaches, including live-cell imaging and transcriptomic profiling, reveal that persistence represents a heterogeneous and dynamic continuum. Subpopulations adopt diverse physiological strategies, from complete translational arrest to slowed but residual metabolic activity, while others leverage DNA repair mechanisms such as transcription-coupled repair to survive antibiotic-induced damage.3,4 This variability demonstrates that persistence is not a binary dormant state but a complex adaptive trait, functioning as a bet-hedging strategy that enhances population survival during fluctuating host defences and antimicrobial assaults.5
The host environment exerts powerful influence over persister formation and maintenance. Within macrophages and epithelial cells, pathogens encounter oxidative stresses such as reactive oxygen species (ROS) and peroxynitrite, which induce ATP depletion and protein aggregation, driving bacteria into deeper dormancy with prolonged awakening lags.6,7 For M. tuberculosis, these host-derived pressures contribute to its ability to remain latent for decades and re-emerge after immune suppression, while simultaneously complicating diagnosis and therapy.8 In S. aureus, intracellular persistence explains why many infections relapse despite aggressive antibiotic therapy, as dormant cells hidden within host cells evade killing and reseed infection.2
Environmental cues outside the host further amplify persistence. Heavy metals such as cadmium and mercury, often found in polluted niches, trigger rapid persister formation by disrupting energy metabolism and redox homeostasis, highlighting environmental determinants of clinical outcomes.9 In biofilms, persisters exploit the protective matrix of extracellular DNA and polysaccharides, where slow penetration of antimicrobials and nutrient gradients promote dormancy and survival. These biofilm-associated persisters are central to device-related infections and chronic wounds.10,11 Additionally, quorum-sensing networks coordinate persistence across biofilm communities, underscoring persistence as both a single-cell and population-level phenomenon.12
The clinical ramifications of persisters extend beyond relapse. By surviving antibiotic exposure, persisters act as reservoirs for genetic adaptation, providing the time and population structure required for the emergence of true resistance mutations. This positions persistence as a bridge between tolerance and resistance, complicating antimicrobial stewardship efforts and elevating the urgency of targeting persisters in therapy design.13 Persistent populations also contribute to high treatment failure rates in biofilm- and host-associated infections, even when pathogens are deemed susceptible by conventional laboratory testing.14
Overall, persister cells are increasingly recognized as a multifaceted survival phenotype shaped by bacterial physiology, host-pathogen interactions, and environmental contexts. Structural insights into ribosome hibernation mechanisms and single-cell studies of awakening dynamics exemplify the rapid progress in mechanistic understanding.15,16 Yet, the persistence phenomenon continues to defy therapeutic eradication, reinforcing its status as both a biological puzzle and a critical clinical obstacle in the fight against antimicrobial resistance.
Definitions and conceptual framework
The concept of persistence has long been intertwined with resistance, tolerance, and the viable but non-culturable (VBNC) state, often leading to confusion in both experimental interpretation and clinical translation. Persister cells are a small subpopulation of genetically wild-type bacteria that withstand otherwise lethal antibiotic concentrations by entering altered physiological states, including dormancy and ribosome hibernation. Unlike resistant mutants, their MIC remains unchanged compared with the susceptible majority, yet they survive prolonged exposure and resume growth once treatment is withdrawn.1,4
Antibiotic resistance, in contrast, is permanent and heritable. Mutations or horizontally acquired genes alter drug targets, permeability, or efflux, raising MIC and allowing the entire population to grow under antibiotic pressure.14 Tolerance differs from both: it is a population-wide phenomenon where genetically susceptible cells survive longer drug exposure without MIC elevation, usually through slowed growth or stress adaptations. Ultimately, however, tolerant cells die if antibiotic exposure persists.3,14 Persistence, therefore, is a quantitative subpopulation trait, while tolerance is a population-level property.2
The VBNC state adds further complexity. VBNC cells remain metabolically active but fail to form colonies on routine media. They can persist for extended periods in hostile environments, but unlike persisters, their resuscitation requires specific stimuli such as pyruvate or glutamate. This distinction has led to the proposal that VBNC and persisters exist along a continuum of dormancy depth, differing mainly in culturability and reactivation cues.17
Clarifying these definitions is not merely semantic. Misclassifying persisters as tolerant or resistant cells risks underestimating their clinical role in relapse and chronicity. A rigorous conceptual framework is essential for guiding research into persister specific pathways such as toxin-antitoxin systems, ribosome hibernation, and DNA repair, and for developing therapies aimed at eradicating these elusive populations.5
As summarized in Table 1, these states differ in genetic basis, killing dynamics, and reversibility.
Table (1):
Distinctions among resistance, tolerance, persistence, and Viable but non-culturable states
Parameters |
Resistance pattern |
AMR Tolerance |
Persistence |
VBNC |
|---|---|---|---|---|
Genetic basis |
Stable mutations / acquired factors: ↑ MIC14 |
None: MIC unchanged14 |
None; MIC unchanged2 |
None: MIC unchanged17 |
Affected population |
Entire population14 |
Entire population3 |
Rare subpopulation (1 |
Variable fraction17 |
MIC relative to susceptible |
Elevated14 |
Same as wild-type14 |
Same as wild-type1 |
Not directly measurable by CFU-based MIC; typically inferred as unchanged using viability assays and/or resuscitation-based readouts.17 |
Killing dynamics |
Survive/grow at high drug levels14 |
Delayed but eventual killing3 |
Biphasic killing with persister tail1 |
Loss of culturability with retained viability: killing dynamics assessed using viability/metabolic assays and/or resuscitation rather than CFU17 |
Growth under drug |
Growth continues14 |
Survival without growth: eventual death14 |
No growth: resume after withdrawal5 |
Active but no colony formation17 |
Reversibility |
Permanent genetic change14 |
Reversible physiological state14 |
Reversible phenotypic state4 |
Reversible with specific resuscitation cues (e.g., pyruvate)17 |
Note: For VBNC cells, standard MIC and time-kill metrics based on turbidity/CFU are not directly applicable because VBNC cells do not form colonies on routine media. Therefore, susceptibility and killing dynamics are typically inferred using non-culture viability assays (e.g., membrane integrity/metabolic indicators) and/or resuscitation based recovery under defined cues.
Operationally, resistance is defined by a stable increase in MIC and sustained growth at antibiotic concentrations that inhibit susceptible cells, typically due to heritable genetic change. Tolerance is defined by a slower killing rate at concentrations above the MIC without an MIC shift and generally involves the bulk population. Persistence is a special case of tolerance in which only a small phenotypically distinct subpopulation survives prolonged exposure, producing a biphasic time-kill-curve survivors regain apparent susceptibility upon regrowth.2,4
Triggers of dormancy and persister formation
The emergence of persister cells is strongly influenced by specific triggers that remodel bacterial physiology in response to antibiotics, host immunity, and environmental conditions. One of the most consistent triggers is antibiotic stress. Exposure to bactericidal agents such as fluoroquinolones induces DNA damage and activation of SOS repair systems, driving a subpopulation of E. coli into persistence characterized by delayed division and survival under lethal conditions.2 Similarly, β-lactam antibiotics promote activation of toxin antitoxin systems and ribosome hibernation, which collectively enable survival during translational arrest.15
Two broad modes of persistence are commonly described: spontaneous (stochastic) and triggered (induced) persistence. Spontaneous persisters arise during balanced growth before an acute stress, driven by phenotypic noise and occasional entry into low-energy/slow-growth states; they remain a small pre-existing subpopulation that seeds the characteristic ‘persister tail’ upon antibiotic challenge. In contrast, triggered persisters form after stress onset (e.g., nutrient limitation/starvation, oxidative/nitrosative stress, host intracellular cues, or antibiotic-mediated damage) through regulatory programs such as the stringent response, toxin-antitoxin activation, and ribosome hibernation. Importantly, both modes can coexist within the same population, contributing to heterogeneous dormancy depth, variable awakening lag times, and relapse potential.11,14
In addition to antibiotics, the host environment is a major source of persister inducing signals. Within macrophages, bacteria encounter reactive oxygen and nitrogen species that collapse ATP pools, generate protein aggregates, and force cells into dormant states with extended awakening times. This has been observed in Mycobacterium tuberculosis during latent infection, where oxidative stress and nutrient limitation sustain non-replicating persistence for decades.8 Likewise, in Staphylococcus aureus, nitric oxide exposure restricts respiration and enhances intracellular persistence, permitting bacteria to survive inside host cells even after aggressive antibiotic treatment.6,7
Environmental stresses also serve as powerful triggers of persistence. Nutrient starvation activates the stringent response through (p)ppGpp accumulation, which globally represses biosynthesis and primes cells for multidrug tolerance.14 Recent work has further highlighted that environmental pollutants, including cadmium and mercury, can significantly increase persister frequency by disrupting redox homeostasis and energy metabolism. These findings suggest that anthropogenic factors in clinical or ecological settings may indirectly worsen treatment outcomes.9
Persistence is further promoted in the biofilm lifestyle, where structural and chemical heterogeneity create favourable conditions. Within biofilms, gradients of oxygen and nutrients generate slow growing subpopulations, while quorum sensing systems coordinate stress responses that enhance survival. Importantly, the biofilm interior typically experiences sustained oxygen/nutrient limitation and reduced growth, which can itself trigger dormancy programs and generate persister like phenotypes even before antibiotic exposure. Because antimicrobials often penetrate unevenly, deep layers may encounter sublethal concentrations or minimal exposure; therefore, survivors in the interior can reflect a mixture of pre-existing (spontaneous) persisters and starvation/gradient-triggered dormant cells, rather than being solely antibiotic induced. Thus, biofilm-associated persistence is best interpreted as a composite outcome shaped primarily by microenvironmental gradients and limited drug penetration.10,11 Persisters embedded in extracellular polymeric substances resist drug penetration and later reseed biofilm growth after treatment, a phenomenon documented in device-associated and wound infections.10,11
Together, these findings demonstrate that persister formation is a multifactorial process shaped by antibiotics, host-derived pressures, environmental challenges, and community level interactions. Recognizing these triggers provides the foundation for designing therapeutic strategies that simultaneously target bacterial physiology and ecological niches, reducing the likelihood of relapse in persistent infections.
Molecular mechanisms underlying persistence
Bacterial persistence emerges from coordinated physiological programs that reduce vulnerability to antibiotics without altering genetic susceptibility. Recent work converges on five mechanistic axes: the stringent response (ppGpp), toxin-antitoxin (TA) and regulatory sRNAs, ribosome hibernation and translational control, DNA-damage responses and repair, and proteostasis/energy remodeling that deepens dormancy. These programs interact to produce heterogeneous cell states that survive bactericidal exposure and later resume growth, explaining biphasic killing and relapse in diverse pathogens.1
The stringent response: (p)ppGpp as a master switch
Nutrient limitation and stress elevate (p)ppGpp via RelA/SpoT enzymes, reprogramming transcription and metabolism toward survival at the expense of growth. Elevated (p)ppGpp suppresses rRNA synthesis, down-shifts biosynthetic flux, and promotes phenotypes with reduced antibiotic target engagement, thereby increasing survival during drug exposure.18 Across organisms, ppGpp-dependent throttling of translation and replication provides a rapid route into low-activity states that mimic dormancy while maintaining viability, making the stringent response a leading target for anti-persister discovery.19
Toxin-antitoxin modules and regulatory sRNAs
TA systems remodel translation, DNA replication, and membrane potential to generate drug-insensitive physiologies. In Staphylococcus aureus, the type I antitoxin SprF1 binds ribosomes to attenuate translation, elevating persister formation and linking sRNA-mediated regulation to survival under antibiotics.20 Broader TA network wiring uncovered by RNA interactome mapping in multidrug-resistant S. aureus suggests extensive cross-talk among sRNAs and TA loci, offering multiple entry points into dormancy and phenotypic tolerance.21 Mechanistic syntheses emphasize that TA effects often converge on translational slowdown, membrane depolarization, or ATP depression changes that buffer lethal antibiotic target reactions without raising MICs.
Ribosome hibernation and translational control
Translational arrest is a core feature of persistent states. Hibernation factors (HPF, RMF and relatives) dimerize or inactivate 70S ribosomes, curtailing translation and shielding ribosomes from damage during stress. Structural and mechanistic analyses across bacteria, including mycobacteria, reveal how hibernated ribosomes stabilize dormancy and enable rapid restart upon stress relief.22 Discovery of a new family of hibernation factors expands the molecular toolkit by which bacteria protect ribosomes in low energy states, underscoring translational quiescence as a conserved persistence strategy.16 Cryo-EM work visualizing the mycobacterial 70S in a hibernated conformation provides structural rationale for reduced drug susceptibility during dormancy and identifies surfaces that could be pharmacologically targeted to prevent or reverse hibernation.23 Ribosome hibernation is typically triggered during nutrient limitation/stationary phase and energy stress (low ATP), and is frequently coordinated with global stress signaling such as the stringent response (p)ppGpp. Under these conditions, bacteria deploy dedicated hibernation factors (e.g., RMF and HPF, and in some species RaiA/YfiA-family proteins) that bind to the 70S ribosome to block mRNA/tRNA access and/or promote formation of inactive 100S ribosome dimers, thereby stabilizing an inactive translational state and protecting ribosomes during prolonged stress. When stress is relieved, these complexes dissociate and translation restarts, enabling persister awakening and regrowth.11,14,23
DNA damage responses, repair pathways, and awakening lags
Bactericidal antibiotics often create drug-target lesions (e.g., fluoroquinolone-stabilized cleavage complexes) and oxidative DNA damage; persisters survive in part by modulating damage processing and repair. Genome wide mapping of quinolone stabilized lesions in E. coli delineates the stress landscape that selects for slow growing survivors.24 Single cell studies show that transcription coupled repair (TCR/NER) capacity shapes awakening lags and survival heterogeneity, providing a mechanistic bridge from transient tolerance to mutational trajectories after recovery.3 In Acinetobacter baumannii, fluoroquinolones and β-lactams induce TCR/NER programs that blunt killing and favor persistence, highlighting DNA repair as a cross-antibiotic survival module.25
Proteostasis, ATP depletion, and dormancy depth
Protein aggregation and ATP collapse correlate with “dormancy depth”, delaying resuscitation and increasing tolerance to antibiotic damage while retaining viability. Conceptual and experimental syntheses identify protein aggregation as both a marker and mediator of persistent states, linking proteostasis stress to translational quiescence and VBNC state transitions.26 In intracellular S. aureus, host derived oxidative stress drives ATP depletion and protein aggregation, pushing bacteria into deep dormancy with long awakening times mechanistic changes that explain survival during therapy and relapse after withdrawal.7 These proteostasis-energy couplings provide actionable levers for therapies that either wake and kill or kill-in-sleep by targeting aggregated proteomes or collapsing protective chaperone circuits.
Population heterogeneity from single-cell physiology
Persistence is not a single, uniform dormant condition but rather a spectrum of physiological states. Time-lapse imaging and lineage tracking studies have shown that bacteria can follow multiple paths to survive antibiotic stress: some persister cells already exist within the population as slow growing or low translation variants, while others are newly induced when stress begins. Many of these cells later display distinct patterns of regrowth once the antibiotic is removed.4 During awakening, coordinated resuscitation events and unequal distribution of cellular damage between daughter cells cause differences in growth rate and drug susceptibility, helping to maintain phenotypic diversity even after treatment.15 These observations explain why bacterial populations show variable killing curves and why not all persisters are completely inactive some remain in partially active metabolic states that reduce drug target engagement and support survival.5
Pathogen-specific contexts integrate these modules
Mechanistic axes are redeployed within host and biofilm environments. In M. tuberculosis, oxidative damage, delayed replication, and subcellular localization modulate both drug access and dormancy programs tied to ribosome control and energy limitation, aligning with clinical phenotypes of latent persistence and diagnostic escape.2,8 In Pseudomonas aeruginosa, persister formation occurs in both planktonic and biofilm states and is reinforced by matrix-mediated diffusion barriers and QS-linked stress modules that throttle growth and translation.10,27 These pathogen-specific deployments of universal modules explain why seemingly unrelated stresses antibiotics, immunity, and environment converge on similar dormant physiologies.11
Therapeutic implications from mechanism
Mechanistic clarity is already guiding anti-persister strategies. Metabolic adjuvants and controlled stressors can transiently restore antibiotic target engagement, e.g. potentiating aminoglycosides by elevating uptake and proton motive force, or even heat-shock preconditioning to break tolerance in Gram-negatives.11 Ribosome-centric approaches target hibernation or exploit antibiotics with activity in low-metabolic cells; recent data show tetracycline-class agents like eravacycline can accumulate in dormant P. aeruginosa persisters and kill during wake-up windows.28 Protease activation strategies that reprogram ClpP (ADEP-like concepts and newer selective activators) aim to liquefy essential proteomes independent of growth, a “kill-in-sleep” tactic directly informed by proteostasis-dormancy coupling.29,30 These illustrations underscore how each mechanistic axis can be turned into a therapeutic lever when combined with precise timing (awakening windows) or adjuvant control of metabolism.1
Synthesis
Mechanisms of persistence are deeply interconnected: ppGpp throttles growth and biases cells toward TA-mediated translational arrest hibernation factors lock ribosomes; DNA-repair capacity and proteostasis set awakening lags and population heterogeneity ensures that not all cells follow the same route into survival. This network view explains robustness of persistence across species and environments and clarifies why multi-pronged interventions (metabolic potentiation + ribosome/repair targeting) are most promising (Figure 1).14
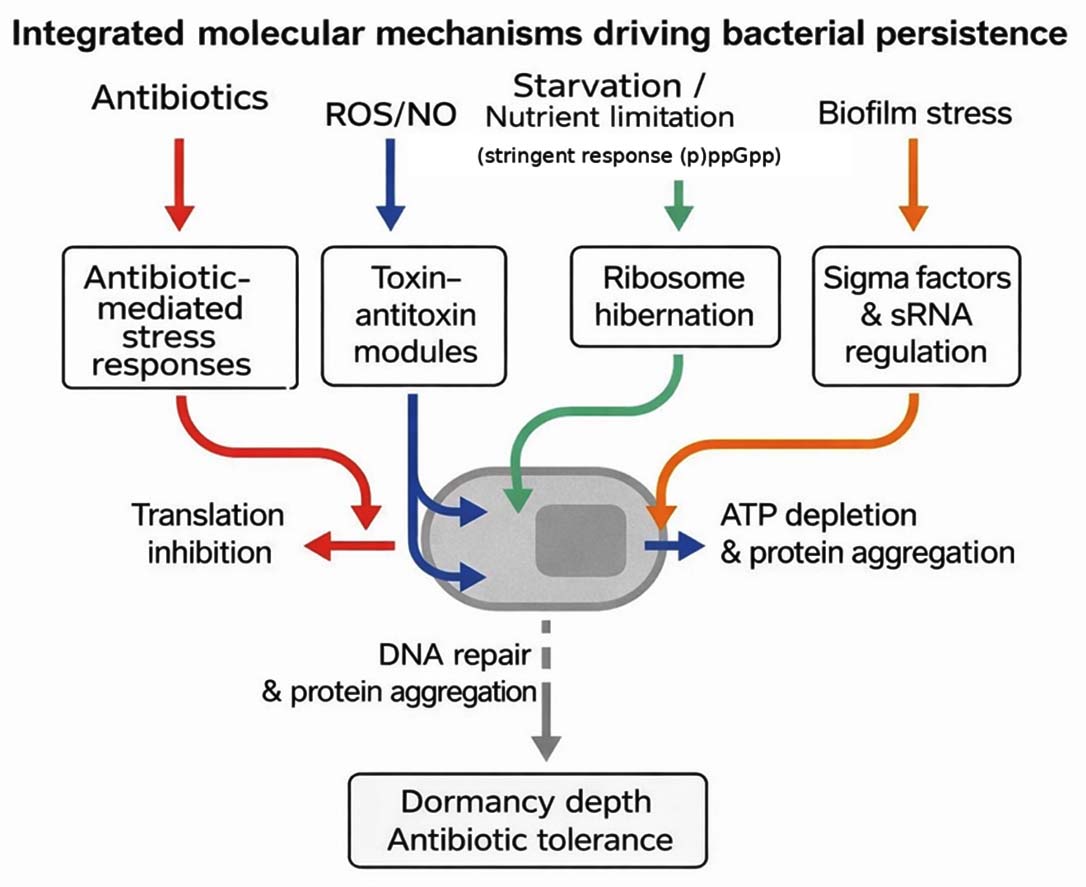
Figure 1. Integrated molecular mechanisms of bacterial persistence
Awakening, reactivation and clinical implications
The transition from dormancy back to active growth termed persister awakening or resuscitation is a critical yet underexplored phase that determines infection relapse. While the formation of persisters is tightly regulated, their awakening is largely stochastic and energy dependent. Single-cell studies reveal that only a fraction of dormant bacteria resume growth rapidly after antibiotic removal, whereas others remain in extended lag states due to differential ATP levels and proteostasis recovery.4
ATP restoration serves as the first checkpoint for awakening. Antibiotic-tolerant cells typically exhibit collapsed energy pools; upon nutrient availability or relief from oxidative stress, glycolytic flux resumes and drives translational restart. In Staphylococcus aureus and Mycobacterium tuberculosis, restoration of intracellular ATP correlates with reactivation of ribosomal function and synthesis of key chaperones.8 This energy burst reactivates translation machinery and reinitiates growth, explaining the heterogeneous regrowth kinetics observed in persister populations.15
Protein disaggregation and proteostasis recovery represent another critical determinant of resuscitation. Dormant persisters accumulate aggregated proteins during stress, which act as “molecular brakes” on growth. Their clearance by ATP-dependent chaperones such as ClpB and DnaK initiates a cascade that restores metabolic flux and division.26 The balance between aggregated and refolded proteomes defines dormancy depth and timing of awakening.
Ribosome reactivation follows proteostasis repair. Hibernation-promoting factors (HPF, RMF) that previously dimerized 70S ribosomes dissociate upon ATP recovery, allowing active translation to resume.16 This sequence energy gain, protein repair, translational restart marks the physiological hallmark of persister awakening.
Environmental and host factors play a crucial role in the reactivation of dormant cells. The availability of nutrients, reintroduction of oxygen, or reduction in nitric oxide levels can stimulate metabolic recovery. In macrophage infection models, for instance, simply withdrawing nitric oxide and restoring redox balance has been shown to awaken intracellular Staphylococcus aureus, illustrating how the host microenvironment directly affects both bacterial persistence and the likelihood of relapse.6
Moreover, differences among individual persister cells lead to asynchronous reactivation, ensuring that not all cells awaken simultaneously. Single cell lineage studies reveal that awakening times can vary considerably, even under identical conditions, resulting in staggered re-establishment of infection sites and contributing to recurrent treatment failure.2
Together, these findings demonstrate that persister awakening is an active, regulated process involving coordinated recovery of energy metabolism, protein homeostasis, and translational capacity. Understanding these transitions provides essential insight for designing “wake-and-kill” strategies that exploit transient windows of vulnerability during resuscitation.
Therapeutic strategies against persisters
Eradicating persister cells requires strategies that go beyond conventional antibiotic therapy. These strategies are summarized in Table 2. Since persisters survive without genetic resistance, current research focuses on approaches that either reactivate dormant cells (“wake-and-kill”) or eliminate them during dormancy (“kill-in-sleep”).
Table (2):
Emerging therapeutic strategies against persister cells
Strategy |
Mechanism |
Example/Agent |
|---|---|---|
Wake-and-kill |
Reactivate metabolism to restore antibiotic sensitivity.31 |
Mannitol, pyruvate, fructose + aminoglycosides |
Kill-in-sleep |
Attack dormant proteomes via ribosome/protease targeting.29 |
Eravacycline; ClpP activators (ADEP, CRMs) |
Host-directed therapy |
Modulate macrophage redox/autophagy: limit niches.8 |
Vitamin D analogs; autophagy enhancers |
Biofilm disruption |
Degrade EPS/eDNA: inhibit quorum sensing.10 |
DNase I; dispersin B: QS inhibitors |
Regulatory inhibition |
Block ppGpp, SOS, or TA responses.25 |
RelA/SpoT inhibitors: TA blockers |
The “wake-and-kill” concept exploits transient windows of metabolic reactivation. Metabolic adjuvants such as mannitol, fructose, or pyruvate restore proton motive force and potentiate aminoglycoside uptake, enabling eradication of otherwise tolerant cells.31 Similarly, energy-boosting strategies that enhance ATP production trigger ribosomal activity and sensitize persisters to bactericidal antibiotics. This approach has shown promise in Pseudomonas aeruginosa and Staphylococcus aureus, where metabolic activation shortens lag phases and increases antibiotic susceptibility.11
Conversely, “kill-in-sleep” tactics target dormant physiology directly. One major direction involves ribosome-targeted drugs such as eravacycline or omadacycline that maintain activity in metabolically inactive cells, penetrating biofilms and inhibiting hibernating ribosomes.28 Another promising class comprises protease-activating agents that dysregulate ClpP protease activity, degrading essential proteins irrespective of growth state. Advanced ADEP derivatives and novel ClpP activators achieve rapid sterilization of persistent S. aureus and Enterococcus populations.29,30
Host-directed therapies (HDTs) are emerging as complementary tools. By modulating macrophage redox balance and boosting autophagic clearance, HDTs limit intracellular niches that foster persistence.8 Combined antibiotic HDT regimens are being explored to disrupt persisters that survive inside immune cells.
Biofilm-disrupting agents offer another crucial dimension. Enzymatic dispersal of extracellular polymeric substances using DNases or dispersin B exposes embedded persisters, improving antibiotic penetration. In clinical models, quorum-sensing inhibitors and matrix-degrading enzymes have reduced tolerance and relapse frequency.10
Finally, anti-stress and repair inhibitors that block SOS, ppGpp, or TA-mediated responses show potential to suppress persister formation altogether. Compounds targeting RelA-SpoT or antitoxin-toxin equilibria are under active investigation, representing preventive avenues against persistence.14,25
Together, these therapeutic frameworks emphasize that persisters can be defeated through metabolic reawakening, translational targeting, proteostasis collapse, and ecological disruption. Combining multiple strategies tailored to pathogen lifestyle planktonic, intracellular, or biofilm remains the most rational path toward clinical clearance.
Challenges and knowledge gaps
Persister biology still struggles with definition and measurement. Labs often mix up persistence with tolerance or VBNC, and kill-curve protocols or MIC-neutral survival metrics are reported inconsistently; this blurs mechanistic interpretation and the link to outcomes, underscoring the need for standardized, phenotype-anchored assays and reporting norms.14
A second barrier is heterogeneity at single-cell resolution. Time-lapse and lineage-tracking consistently reveal broad distributions of entry routes, lag times, and regrowth behaviours after drug withdrawal, yet translating these dynamics into clinically meaningful predictions inside host tissues or biofilms remains difficult.4,15 This gap limits our ability to time therapies to transient vulnerability windows during awakening.
Mechanistically, causality among coupled modules is not fully resolved. Protein aggregation, ATP collapse, ribosome hibernation, and DNA-repair programs frequently co-occur, but their ordering and relative necessity appear context-dependent across species and stresses.26 Recent structural work clarifying hibernated ribosome states and single-cell data on transcription-coupled repair during recovery help place pieces of the puzzle, yet actionable choke points that generalize across niches are still being mapped.3,16
Model systems also underrepresent hard clinical niches. Biofilms impose diffusion barriers and metabolic gradients that favour dormancy, while intracellular reservoirs experience host-derived oxidants and nutrient limitation; both environments select for persistence and complicate antibiotic access.8,10,11 On the diagnostic side, culture-based tests miss non-growing survivors, and molecular proxies for dormancy depth or imminent resuscitation such as damage-repair signatures require validation before clinical deployment.
Therapeutically, single agents rarely sterilize persisters. Rational combinations that couple metabolic reactivation with bactericidal antibiotics, or that pair ribosome/protease-targeted agents with biofilm dispersal, look promising but will require dosing schedules synchronized to awakening kinetics and niche ecology.28,31 Looking ahead, priorities include consensus standards for persistence assays, spatial single-cell multi-omics in tissue-like models, validated biomarkers that report dormancy depth in situ, and adaptive combination regimens that exploit short resuscitation windows while dismantling biofilm and intracellular shelters.11,15
Graphical summary of bacterial persistence and therapeutic strategies
Bacterial persistence represents a flexible survival behaviour that lets a few bacterial cells tolerate otherwise lethal antibiotic doses without becoming genetically resistant. This process usually begins when the cells face stress from antibiotics, the host immune system, or their own slow-growing biofilm environment. Under these conditions, the bacterial metabolism slows dramatically, and several molecular safety nets start working together. Molecules such as (p)ppGpp reduce energy-consuming pathways, toxin antitoxin systems pause translation, and ribosome hibernation protects the protein-making machinery. DNA-repair enzymes and proteostasis networks help the cell stay alive even when energy levels drop.
When stress eases, these dormant persisters do not all recover at once. Some cells regain ATP quickly, dissolve protein aggregates, and restart translation, while others remain inactive for long periods. This uneven “awakening” explains why infections can relapse after apparently successful antibiotic therapy. Recognising these recovery steps has inspired new treatment ideas re-energising cells before drug exposure (“wake-and-kill”), striking them while still inactive (“kill-in-sleep”), altering host defences, or dismantling biofilms to expose hidden survivors.
Figure 2 summarises this overall framework, linking the formation of dormant persisters, their molecular maintenance systems, and the therapeutic approaches now being explored to prevent relapse and resistance.
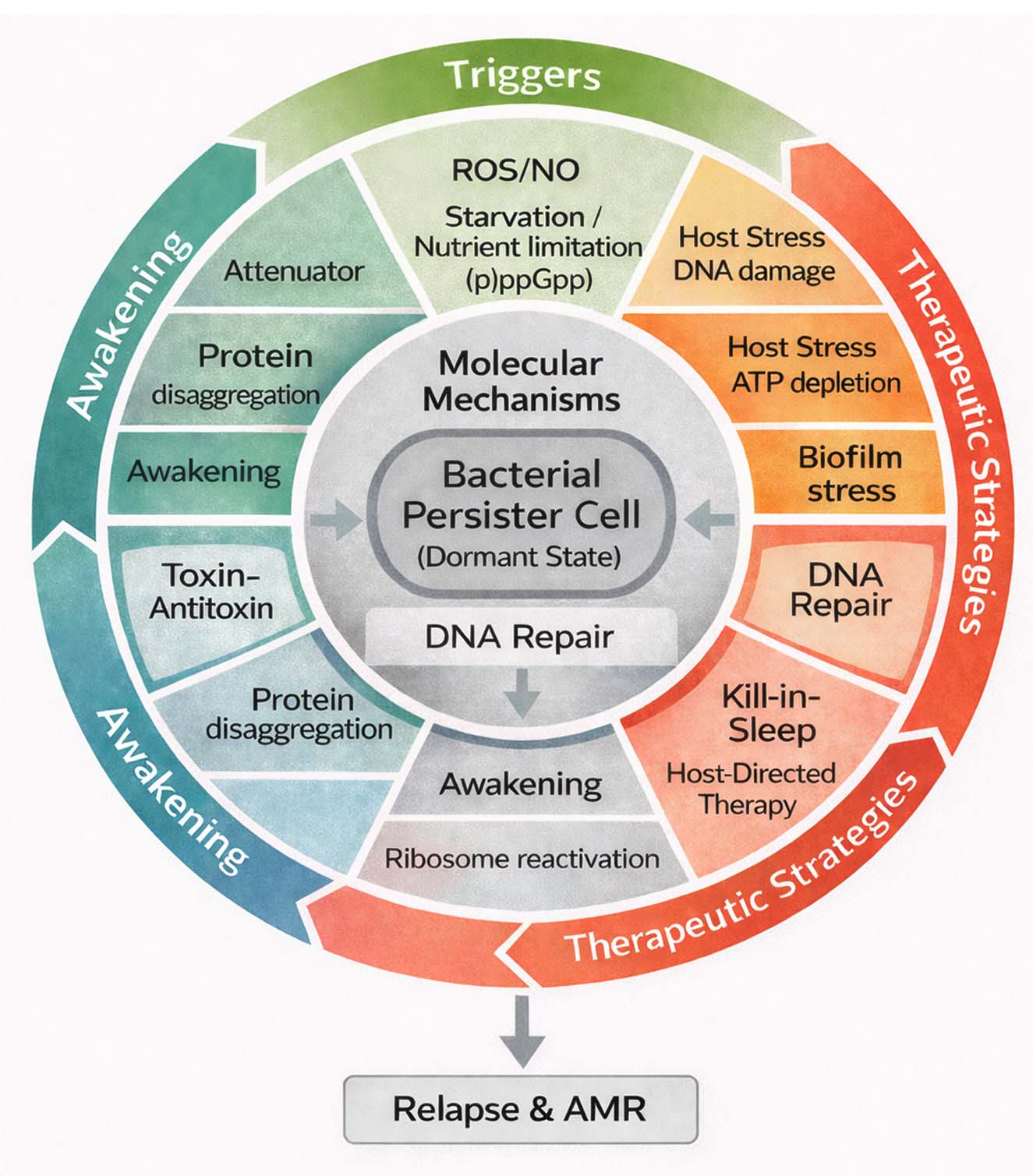
Figure 2. Integrated overview of bacterial persistence and therapeutic strategies
Limitations and advantages
Although research on bacterial persisters has advanced quickly, several gaps remain between laboratory findings and what actually happens in patients. Many studies depend on simplified in vitro models that do not capture the constant nutrient changes, immune pressure, or micro-niches found inside the body. As a result, results that seem clear in the lab often behave differently in living hosts. Distinguishing true persisters from other non-growing forms such as VBNC cells is also difficult, because both look similar under standard culture conditions. Reliable markers or rapid tests that identify persisters during infection are still missing, so clinicians usually detect them only after relapse occurs.
Experimental therapies that target persisters like metabolic activators or ClpP protease stimulators show promise but are still at an early stage. Their safety, dosing, and effects on normal microbiota need more work. Despite these limits, the field has gained strong advantages in the past few years. Modern single-cell imaging and transcriptomic tools now let researchers follow individual cells as they enter or leave dormancy. Structural studies of ribosome hibernation and toxin-antitoxin modules have revealed new drug targets. Together, these advances have given a clearer picture of how persistence works and opened the door to combination treatments that awaken or directly eliminate dormant bacteria.
So, while persister biology still has experimental and clinical hurdles, its integration of molecular and ecological insights is steadily bringing the goal of reliable anti-persister therapy closer to reality.
Persisters are neither passive survivors nor rare anomalies but rather a dynamic subpopulation shaped by ecological, host, and therapeutic pressures. The past five years have consolidated evidence that persistence is underpinned by converging regulatory programs ppGpp signaling, toxin-antitoxin balance, ribosome hibernation, and proteostasis that coordinate survival during stress. Yet many mechanistic relationships remain correlative, and our models often fail to capture the complexity of host and biofilm niches. Eradication requires more than new antibiotics; it demands adaptive, combination-based strategies that integrate metabolic activation, protease or ribosome targeting, host-directed modulation, and biofilm disruption. Moving forward, standardization of assays, single-cell and multi-omics integration, and validated biomarkers of dormancy depth will be pivotal. By bridging mechanistic insight with ecological realism, persister research can transition from conceptual mystery to translational opportunity, offering a path to more durable infection control.
ACKNOWLEDGMENTS
None.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
AUTHORS’ CONTRIBUTION
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
FUNDING
None.
DATA AVAILABILITY
Not applicable.
ETHICS STATEMENT
Not applicable.
- Niu H, Gu J, Zhang Y. Bacterial persisters: molecular mechanisms and therapeutic development. Signal Transduct Target Ther. 2024;9(1):174.
Crossref - Peyrusson F, Varet H, Nguyen TK, et al. Intracellular Staphylococcus aureus persisters upon antibiotic exposure. Nat Commun. 2020;11(1):2200.
Crossref - Wilmaerts D, Focant C, Matthay P, Michiels J. Transcription-coupled DNA repair underlies variation in persister awakening and the emergence of resistance. Cell Rep. 2022;38(9):110427.
Crossref - Umetani M, Fujisawa M, Okura R, et al. Observation of persister cell histories reveals diverse modes of survival in antibiotic persistence. eLife. 2025;14:e79517.
Crossref - Zou J, Peng B, Qu J, Zheng J. Are Bacterial Persisters Dormant Cells Only? Front Microbiol. 2022;12:708580.
Crossref - Beam JE, Wagner NJ, Shook JC, et al. Macrophage-Produced Peroxynitrite Induces Antibiotic Tolerance and Supersedes Intrinsic Mechanisms of Persister Formation. Infect Immun. 2021;89(10):e00286-21.
Crossref - Peyrusson F, Nguyen TK, Najdovski T, Van Bambeke F. Host Cell Oxidative Stress Induces Dormant Staphylococcus aureus Persisters. Microbiol Spectr. 20223;10(1):e02313-21.
Crossref - Saito K, Mishra S, Warrier T, et al. Oxidative damage and delayed replication allow viable Mycobacterium tuberculosis to go undetected. Sci Transl Med. 2021;13(621):eabg2612.
Crossref - Baek S, Seo J, Yun T, et al. Heavy metals promote the formation of multidrug-tolerant Staphylococcus aureus and Escherichia coli persisters. Ecotoxicol Environ Saf. 2025;293:118014.
Crossref - Backhaus J, Kann S, Hahn A, et al. Clustering of Gastrointestinal Microorganisms in Human Stool Samples from Ghana. Pathogens. 2024;13(7):583.
Crossref - Soares A, Alexandre K, Etienne M. Tolerance and Persistence of Pseudomonas aeruginosa in Biofilms Exposed to Antibiotics: Molecular Mechanisms, Antibiotic Strategies and Therapeutic Perspectives. Front Microbiol. 2020;11:2057.
Crossref - Personnic N, Striednig B, Hilbi H. Quorum sensing controls persistence, resuscitation, and virulence of Legionella subpopulations in biofilms. ISME J. 2021;15(1):196-210.
Crossref - Kunnath AP, Suodha Suoodh M, Chellappan DK, Chellian J, Palaniveloo K. Bacterial Persister Cells and Development of Antibiotic Resistance in Chronic Infections: An Update. Br J Biomed Sci. 2024;81:12958.
Crossref - Bollen C, Louwagie E, Verstraeten N, Michiels J, Ruelens P. Environmental, mechanistic and evolutionary landscape of antibiotic persistence. EMBO Rep. 2023;24(8):e57309.
Crossref - Fang X, Allison KR. Resuscitation dynamics reveal persister partitioning after antibiotic treatment. Mol Syst Biol. 2023;19(4):e11320.
Crossref - Helena-Bueno K, Rybak MY, Ekemezie CL, et al. A new family of bacterial ribosome hibernation factors. Nature. 2024;626(8001):1125-1132.
Crossref - Dewachter L, Bollen C, Wilmaerts D, et al. The Dynamic Transition of Persistence toward the Viable but Nonculturable State during Stationary Phase Is Driven by Protein Aggregation. mBio. 2021;12(4):e00703-21.
Crossref - Pacios O, Blasco L, Bleriot I, et al. (p)ppGpp and Its Role in Bacterial Persistence: New Challenges. Antimicrob Agents Chemother. 2020;64(10):e01283-20.
Crossref - Ranea-Robles P, Violante S, Argmann C, et al. Murine deficiency of peroxisomal l-bifunctional protein (EHHADH) causes medium-chain 3-hydroxydicarboxylic aciduria and perturbs hepatic cholesterol homeostasis. Cell Mol Life Sci. 2021;78(14):5631-5646.
Crossref - Pinel-Marie ML, Brielle R, Riffaud C, Germain-Amiot N, Polacek N, Felden B. RNA antitoxin SprF1 binds ribosomes to attenuate translation and promote persister cell formation in Staphylococcus aureus. Nat Microbiol. 2021;6(2):209-220.
Crossref - Mediati DG, Wong JL, Gao W, et al. RNase III-CLASH of multi-drug resistant Staphylococcus aureus reveals a regulatory mRNA 3’UTR required for intermediate vancomycin resistance. Nat Commun. 2022;13(1):3558.
Crossref - Ueta M, Ohniwa RL, Yoshida H, Maki Y, Wada C, Wada A. Role of HPF (Hibernation Promoting Factor) in Translational Activity in Escherichia coli. J Biochem (Tokyo). 2008;143(3):425-433.
Crossref - Kumar N, Sharma S, Kaushal PS. Cryo- EM structure of the mycobacterial 70S ribosome in complex with ribosome hibernation promotion factor RafH. Nat Commun. 2024;15(1):638.
Crossref - Tang J, Brynildsen MP. Genome-wide mapping of fluoroquinolone-stabilized DNA gyrase cleavage sites displays drug specific effects that correlate with bacterial persistence. Nucleic Acids Res. 2023;51(3):1208-1228.
Crossref - Mattiello SP, Barth VC, Scaria J, Ferreira CAS, Oliveira SD. Fluoroquinolone and beta-lactam antimicrobials induce different transcriptome profiles in Salmonella enterica persister cells. Sci Rep. 2023;13(1):18696.
Crossref - Bollen C, Dewachter L, Michiels J. Protein Aggregation as a Bacterial Strategy to Survive Antibiotic Treatment. Front Mol Biosci. 2021;8:669664.
Crossref - Patel H, Buchad H, Gajjar D. Pseudomonas aeruginosa persister cell formation upon antibiotic exposure in planktonic and biofilm state. Sci Rep. 2022;12(1):16151.
Crossref - Roy S, Cakmak ZS, Mahmoud S, Sadeghzadeh M, Wang G, Ren D. Eradication of Pseudomonas aeruginosa Persister Cells by Eravacycline. ACS Infect Dis. 2024;10(12):4127-4136.
Crossref - Yang Y, Wang Y, Zeng F, Chen Y, Chen Z, Yan F. Ultrasound-visible engineered bacteria for tumor chemo-immunotherapy. Cell Rep Med. 2024;5(5):101512.
Crossref - Brotz-Oesterhelt H, Vorbach A. Reprogramming of the Caseinolytic Protease by ADEP Antibiotics: Molecular Mechanism, Cellular Consequences, Therapeutic Potential. Front Mol Biosci. 2021;8:690902.
Crossref - Lang M, Carvalho A, Baharoglu Z, Mazel D. Aminoglycoside uptake, stress, and potentiation in Gram-negative bacteria: new therapies with old molecules. Microbiol Mol Biol Rev. 2023;87(4):e00036-22.
Crossref
© The Author(s) 2026. Open Access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License which permits unrestricted use, sharing, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.