


ISSN: 0973-7510
E-ISSN: 2581-690X
This study analyzed the community of bacteria present on the epidermal layer of Hippocampus barbouri using 16S rRNA amplicon sequencing. A total of 103 Operational Taxonomic Units (OTUs) comprising 61 unique bacterial species were identified, with female samples exhibiting a higher read count (164,844) compared to males (142,525). The predominant bacterial phyla include Pseudomonadota, Bacteroidota, Bacillota, Actinomycetota, and Cyanobacteriota, with Pseudomonadota being prevalent for both samples comprising 50% for females and 53.33% for males. Shewanella baltica was the most abundant species found on female skin, likely due to their increased mobility and exposure to shallow marine waters. Identified bacterial families includes Flavobacteriaceae, Pseudomonadaceae, Moraxellaceae, and Vibrionaceae. The greater bacterial diversity observed in female samples in this study may be attributed to their broader range of movement and environmental interactions. Phylogenetic analysis confirmed species relationships and suggested that seahorse mobility plays a role in shaping skin microbiota. These findings highlight sex-specific differences in microbial composition and emphasize the potential influence of host-microbiome interactions in marine organisms.
Bacteria, Hippocampus barbouri, Seahorse, Species, 16S rRNA
Hippocampus barbouri, commonly known as the Barbour’s seahorse, is a species of seahorse that has garnered interest due to its unique biological and ecological characteristics, including its role in marine ecosystems and its vulnerability to environmental changes. Like other seahorse species, H. barbouri exhibits complex behaviors, including male pregnancy, which sets the species apart from many other marine organisms.1 This reproductive strategy has not only influenced the evolutionary biology of the species but also shaped behavioral patterns, particularly in terms of spatial distribution and social interactions.2 While much attention has been given to their reproductive habits and ecological roles, less is known about the microbial communities associated with their dermal surfaces specially on identification up to the species level.
The skin of marine organisms serves as a critical interface with their environment, often hosting diverse microbial communities that contribute to various physiological functions.3 In seahorses, bacteria in the epidermis are vital for the health and immune system of the host, protecting against pathogenic microorganisms, and facilitating ecological interactions within the marine environment.4 Like other seahorses, its skin features a unique dermal structure, with the presence of flame cone cells which is covered by a mucous cap within its surface. Unlike typical cells, these mucoid caps on the seahorse’s skin support the growth of epiphytic microbes.5 This specialized skin structure is a reservoir for environmental bacteria, which ultimately become part of the seahorse’s dermal microbial communities. These microbial communities, collectively known as the dermal microbiota, are shaped by environmental factors, host physiology, and behavioral traits.6 Recent advances in metagenomic sequencing and bioinformatics have enabled more detailed exploration of these microbial populations, offering insights into their diversity, functional significance, and the factors influencing their composition.7
While microbial communities are commonly studied at the species level, the potential for sex-specific differences in these communities has received relatively little attention. In H. barbouri, as in other seahorse species, male and female individuals exhibit distinct physiological and behavioral traits that may lead to differential microbial exposures. For example, male pregnancy significantly alters the reproductive behavior and physiology of males compared to females, possibly influencing the microbial populations on their skin. Additionally, females in many seahorse species tend to have larger home ranges, more social interactions, and greater mobility within marine ecosystems, all of which could expose them to a wider variety of microbial environments than their male counterparts.8 Exploring sex-specific differences in the dermal microbial communities of H. barbouri is important for understanding the broader implications of host-microbiome interactions. Such differences could reveal how microbial populations are influenced not only by the host species but also by its sex, providing new understandings into the function of microbiota in host health and environmental adaptation. Furthermore, understanding these microbial differences at the species level may offer valuable information for conservation efforts, particularly in managing the health of this vulnerable species in cases of altered environments.
Moreover, this study is particularly significant as fish skin harbors a diverse mucosal microbiota, offering valuable insights into microbial community ecology with potential applications in agriculture and veterinary medicine. It represents a key area for understanding interactions between microbial community members and their influence on fish health, particularly about specific nutrients and microbial species. Genes associated with skin colonization, such as attachment or mucin degradation, must be further explored. Additionally, directly linking skin immunity factors to microbial composition and individual taxa is essential. While a strong foundation has been established, numerous exciting discoveries still await.9
Thus, we evaluate the species-level structure of the epidermal microbial communities associated with male and female H. barbouri, using advanced molecular techniques to detect bacterial species present on the skin of both sexes. By comparing the microbial communities of male and female individuals, this research seeks to uncover potential sex-specific patterns that behavioral and physiological differences may influence. The findings of this study could contribute to a deeper understanding of the role of microbiota in marine organisms, shedding light on the complex interactions between seahorses and their microbial environments, and potentially informing conservation and management strategies for this species.
Study area and sampling collection
Permits were obtained prior to the collection of samples in this study. GP No. 0249-23 was acquired from the National Bureau of Fisheries and Aquatic Resources since bacterial sources is a host which is nearly endangered. Courtesy calls were made to local residents and community officials to inform them of the sampling activity in the area and ensure their awareness and support. Thirty samples (15 male and 15 female) of Hippocampus barbouri (approx. 5-6 g in weight) were obtained from the coral areas on the island of Cantiasay in Surigao del Norte (9°512 343 N 125°362 043 E). Samples were directly transported and processed to the Molecular Systematics and Conservation Genomics Laboratory of the Premier Research Institute of Science and Mathematics (PRISM) in Mindanao State University-Iligan Institute of Technology. Two gentle washes with sterile seawater were performed on seahorse samples to get rid of the debris without harming the skin’s microbial colonies.10 Using a sterile bacterial swab, the dorsolateral surface of the Hippocampus barbouri was scraped to collect the bacteria from the skin mucus.11 For DNA extraction, bacterial colonies isolated from the skin were put on a 2 ml microcentrifuge tube and kept at -65 °C.
DNA extraction, amplification, purification, sequencing and analysis
About 2 ml of the eluted buffer solution containing the bacterial isolate’s total genomic DNA was taken out. The liquid culture was then centrifuged, and the bacterial DNA was isolated using bead-beating from the HiPurA™ Sediments DNA Purification Kit (Vadhani Industrial Estate, Mumbai, India) with slight modifications. The DNA isolates was sent to Macrogen Incorporated in South Korea, for purification and sequencing. The generated reads from Macrogen were comprehensively quality filtered, trimmed, and assembled. Overlapping regions, mismatch, and the paired-end sequences that were present in the sequence assembly were discarded. Additionally, any sequences with unclear base calls was removed. After trimming and assembly, sequence reads were checked using SILVA online software for ribosomal RNA sequence data quality and alignment. This software is also used to obtain several charts necessary to visualize the bacterial composition and the quantity of the relative abundance of the identified bacteria.12 Five essential modules perform data processing: align, quality control, dereplication, clustering, and classification. SILVA Incremental Aligner was used to align each read (SINA SINA v1.2.10 for ARB SVN (revision 21008))13 against the SILVA SSU rRNA SEED and quality controlled.12 Reads that were 2% ambiguous or homopolymers, or shorter than 50 aligned nucleotides, were not included for additional processing. Sequence reads with poor alignment quality and possible contaminants and artifacts were also found and eliminated from further examination. Following the quality control stage, the unique reads were clustered (OTUs) and identified by dereplication. Cd-hit-est (version 3.1.2) was used to do dereplication and clustering.14 Each OTU was categorized using a local nucleotide BLAST search against the non-redundant version of the SILVA SSU Ref dataset.15 The SILVAngs fingerprint and Krona charts assigned the meta group “No Relative” to reads that had no BLAST hits or weak BLAST hits, where the function/(% sequence identity + % alignment coverage)/2″ did not surpass the value of 93. SILVANGS provides taxonomic fingerprints to determine the relative proportions of each bacterial group.16 The phylogenetic inference for each species was conducted by comparing the obtained sequences with those available in an online database (NCBI GenBank) to confirm species identification. To provide strong statistical evidence for the predicted associations, a Maximum Likelihood (ML) tree was built using the aligned sequences and 1000 bootstrap iterations.17
Nucleotide sequence accession number
Amplicon sequence data derived from this study was submitted to the GenBank database at NCBI with BioProject PRJNA1217945 with accession number SRR32211362.
This study analyzed bacterial communities from male and female Hippocampus barbouri skin samples through 16S rRNA metagenomic sequencing. 103 operational taxonomic units (OTUs) were identified, with no sequences rejected (0.00% rejection). This total OTUs have generated 33 distinct families, 44 genera and 61 species. The length of the sequences is ranging from 420 bp to 465 bp, having the average of 428 bp. Percent similarity between individual sample groups was 93%, indicating the generation of high-quality sequences. A total of 307,399 amplicon sequence reads were obtained, with 164,844 reads derived from the female skin samples and 142,525 reads from the male skin samples.
The taxonomic fingerprint reveals that all bacterial group sequences are <100, indicating uniformity across all samples and providing an accurate view of the population with moderate recovery biases accounted for. Using taxonomic analysis, five phyla were found in the V1-V3 areas of the 16S rRNA gene amplicon reads: Pseudomonadota, Bacteroidota, Bacillota, Campylobacterota and Cyanobacteria (Figure 1). Pseudomonadota dominated the female and male skin samples, comprising 50% and 53.33% respectively. This study identified ten bacterial classes. While the overall diversity of bacterial classes was consistent between the two sample groups, some classes represented less than 1% of the total bacterial isolates, limiting their resolution. Gammaproteobacteria was the dominant class in female skin samples, comprising 38.89%, followed by Flavobacteria (16.67%) and Alphaproteobacteria (11.11%). Meanwhile, the dominant class in male skin samples was identified to be class Gammaproteobacteria (26.67%), followed by Bacilli at 20%. Flavobacteriaceae was the most dominant family for female skin samples, representing 20% of the total population. Other prominent families included Moraxellaceae (17.78%), Shewanellaceae (8.89%), Rhodobacteraceae (6.67%), and Aeromonadaceae (6.67%). On the contrary, in the male skin, the Family Bacillaceae was the most abundant family, comprising 13.33% of the total population.
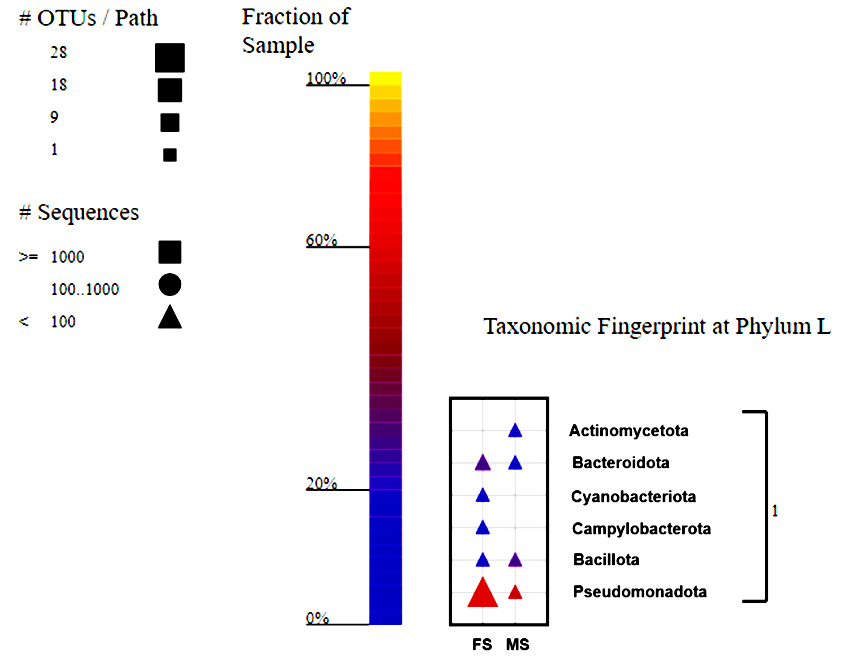
Figure 1. Taxonomy fingerprint at the phylum level of the male and female skin samples of H. barbouri
The generated sequence obtained from female and male skin samples was positioned on a Phylogenetic tree showing the homology of species with closely related bacterial species from an existing online database (Figures 2 and 3). Bacteria isolated from the samples were tagged for OTU number for easy identification. For Figures 2 and 3, the Maximum Likelihood approach, which is based on the Kimura 2-parameter model, was used to infer the evolutionary history of the detected bacterial species. The evolutionary history of the examined taxa is shown by the bootstrap consensus tree that was inferred from 500 replicates.18 Collapsed branches are those that correspond to partitions that were replicated in fewer than 50% of bootstrap replicates. By applying the Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the Maximum Composite Likelihood (MCL) approach, the initial trees for the heuristic search were automatically generated. The branches are accompanied by the percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (500 replicates).19 The topology with the superior log likelihood value was then chosen. In MEGA7, evolutionary analyses were performed.
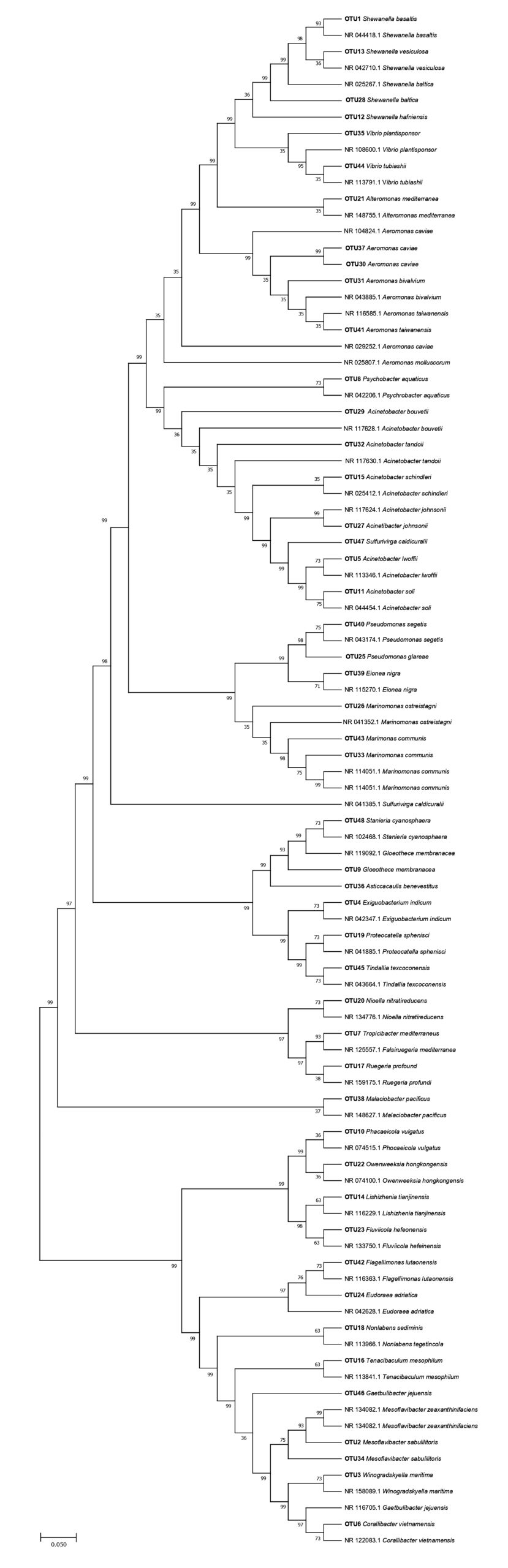
Figure 2. Phylogenetic inference of skin microbial species of female H. barbouri with closely related bacterial species from existing online database using 16S rRNA gene sequences constructed by maximum likelihood analysis. The scale bar of 0.050 represents nucleotide substitution
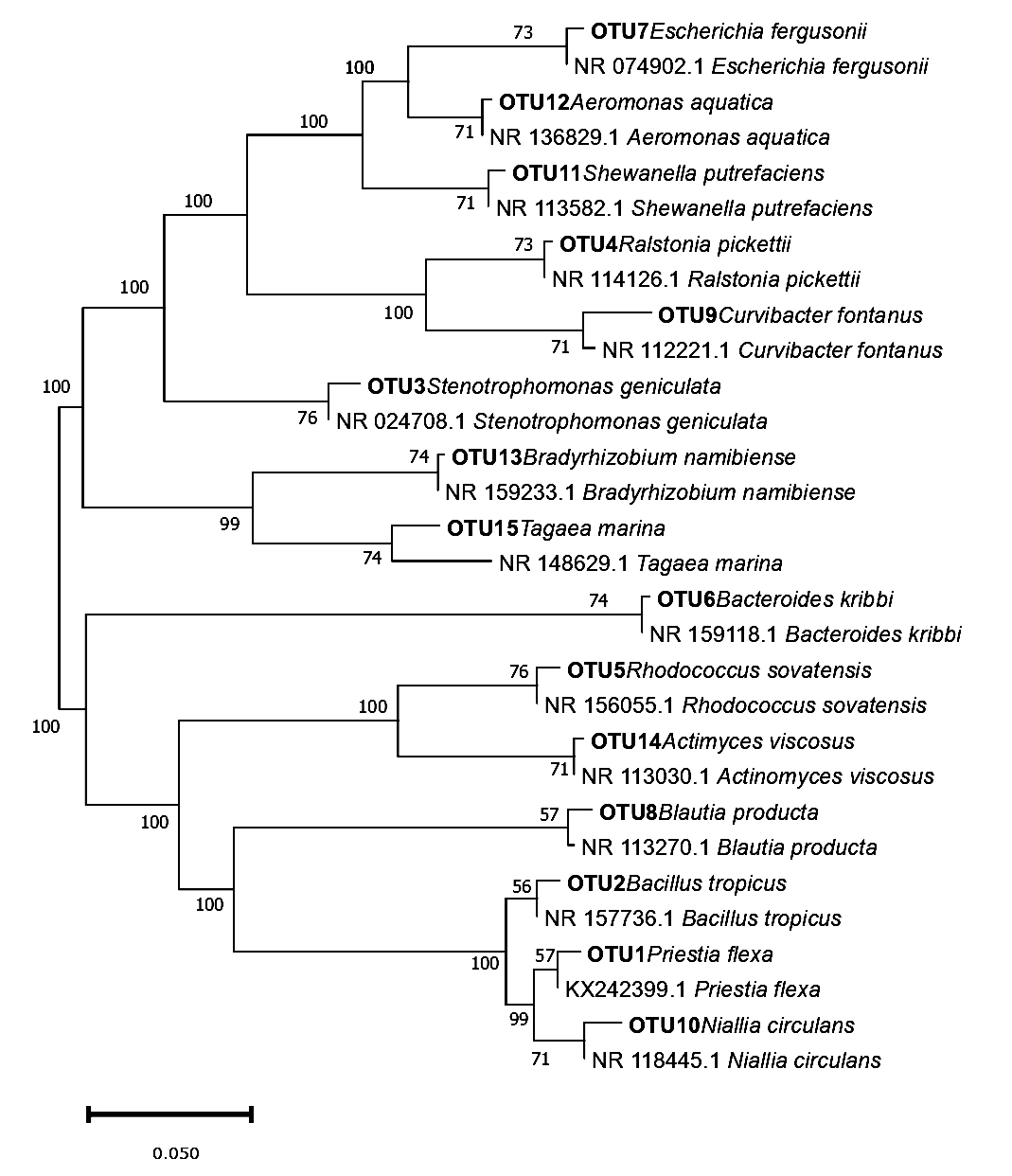
Figure 3. Phylogenetic inference of skin microbial species of male H. barbouri with closely related bacterial species from existing online database using 16S rRNA gene sequences constructed by maximum likelihood analysis. The scale bar of 0.050 represents nucleotide substitution
The tree with the highest log likelihood (-4796.13) is displayed in Figure 2. Evolutionary rate differences between locations were modeled using a discrete Gamma distribution; in five categories, the distribution/parameter equaled 0.9694. At 43.30 percent, the rate variation model permitted some sites to be evolutionarily invariant. The impact of site variation on estimates of transition bias is demonstrated using a gamma-distributed-rates model. After removing the sites with gaps and missing data, the final dataset in this sample’s study comprises 345 positions and 94 nucleotide sequences. Figure 3 shows how evolutionary rate differences between sites with five categories were modeled using a discrete Gamma distribution with a value of 200.30 nucleotide sequences were analyzed. First, second, third, and noncoding codon locations were covered.
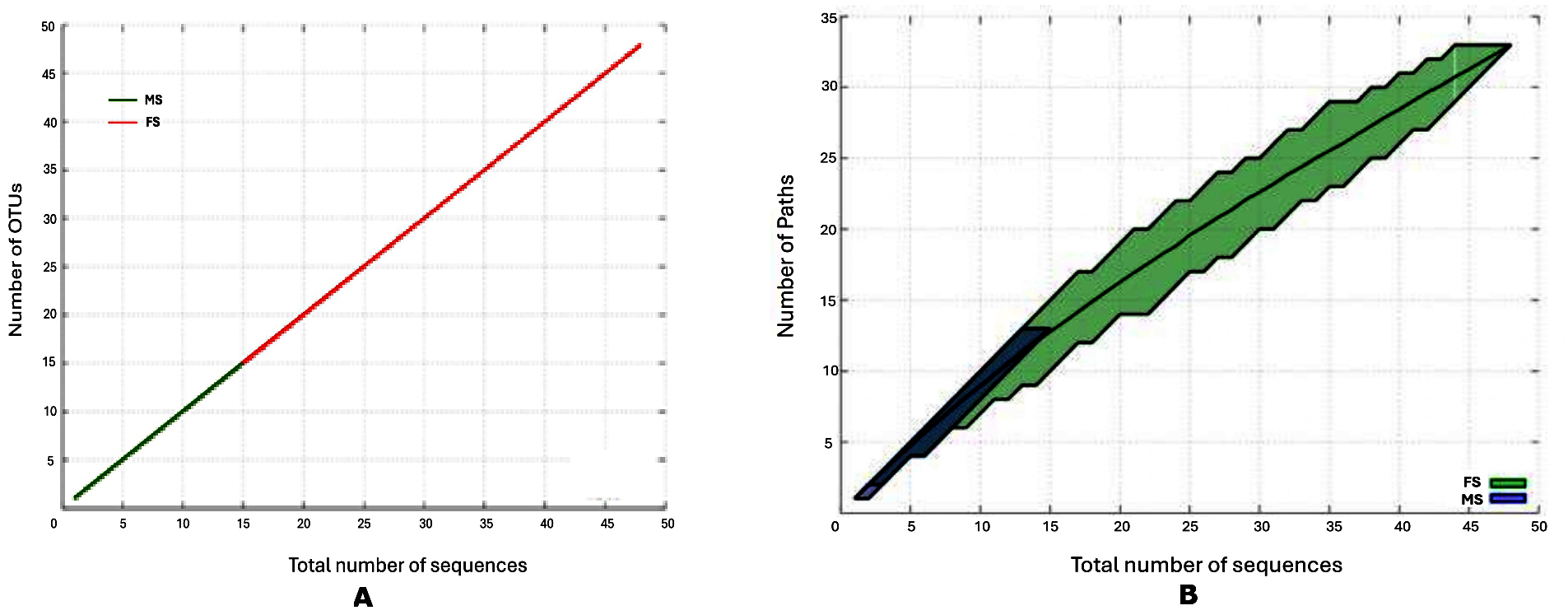
Figure 4. Rarefaction graphs showing the influence of percentage dissimilarity on the number of OTUs (A) and Paths (B) detected
Analysis of the species richness based on OTU count (Figure 4) shows that in the female skin sample, when compared to the male skin sample, the OTU abundance is noticeably higher at high degrees of dissimilarity. The female skin microbiota may include a more varied and heterogeneous microbial community than the male skin sample, as indicated by the noticeably greater OTU abundance seen in the female skin sample at high levels of dissimilarity. This finding is consistent with previous studies, which highlight gender-specific variations in skin microbiota due to differences in skin physiology, hormone levels, and environmental exposures.20 The failure of the species richness curve for both samples to reach a plateau indicates that the sampling depth was insufficient to capture the full extent of microbial diversity. This limitation suggests that additional sampling would be required for a comprehensive comparison of the microbial communities. Despite this limitation, the current data remains valid and meaningful for characterizing the comparative richness between the two samples. Such findings align with previous work emphasizing the importance of sampling depth in microbial diversity studies.21
In this study, Amplicon sequencing analysis using Next-Generation Sequencing (NGS) successfully identified the bacterial groups present with male and female Hippocampus barbouri. This approach enables the detection of both cultivable and uncultivable bacteria in the samples. Results showed that the female skin samples harbored a higher diversity and abundance of microbial flora compared to the male. Higher bacterial diversity in the female H. barbouri may be attributed to its greater mobility within its home range. Females tend to exhibit wider-ranging behaviors, interacting with diverse environments. In contrast, males typically have smaller home ranges, limiting their daily movements. This restricted mobility, a trait observed in other monogamous syngnathid species,22 may reduce exposure to environmental microbial sources, thereby limiting bacterial colonization on the male’s skin. The female’s higher maneuverability, advantageous for activities such as mating and predation, exposes them to a broader range of habitats, including eelgrass beds and coral reefs.23-25 Some studies noted that while females may exhibit less active movement, they spend more time swimming across larger territories.26,27 This behavior increases their exposure to diverse microbial populations. Additionally, a study in 2012 suggested that females occupy a larger proportion of the environment, moving with minimal motion but covering extensive areas.28 These findings suggest that the wider range of interactions and habitats experienced by female H. barbouri contributes to the higher diversity and abundance of bacterial communities observed on their skin.
Shewanella baltica was the most abundant species identified in female skin samples, comprising 41.29% of the total bacterial population. This species is recognized as a natural inhabitant of marine environments and is well-adapted to warmer climates, typically found in shallow temperate waters.29 The abundance of S. baltica on the skin of female Hippocampus barbouri can be attributed to the seahorse’s habitat preferences, as this species of seahorse is commonly found in shallow ocean regions. Additionally, the itinerant nature of female H. barbouri increases their exposure to environmental microbial communities, facilitating the colonization of S. baltica on their skin. This observation supports the notion that the external microbial flora of marine organisms reflects the composition of their surrounding environment.30 The prevalence of S. baltica in shallow marine waters aligns with the behavior and habitat of female H. barbouri, explaining its significant presence in the bacterial population associated with their skin.
On the other hand, genera identified in this study were dominated by Acinetobacter, Shewanella, Aeromonas, Marimonas, Ruegeria, Pseudomonas, Vibrio, Mesoflavibacter belonging to the families Flavobacteriaceae, Pseudomonadaceae, Moraxellaceae, Aeromonadaceae, Vibrionaceae, and Bacillaceae respectively. The predominance of the genus Acinetobacter may be attributed to its metabolic adaptability, as species within this genus are often found in diverse environments, including marine habitats, and are recognized for their roles in organic matter degradation.31 Shewanella and Aeromonas are also commonly associated with aquatic environments and are known for their ability to reduce a wide variety of compounds, making them important contributors to biogeochemical cycling.32 The presence of Ruegeria, a member of the Rhodobacteraceae family, is noteworthy due to its role in symbiotic relationships with marine organisms and its potential contribution to sulfur cycling in aquatic systems.33 Pseudomonas, a genus well-known for its metabolic diversity, likely plays a role in nutrient cycling and may exhibit antimicrobial properties that influence microbial community composition.34 The detection of Vibrio is significant given its dual role as both a symbiotic and pathogenic genus in marine environments. Species within this genus are often associated with aquatic hosts and can impact host health through the production of bioactive compounds.35 Similarly, the identification of members from the family Bacillaceae, including Mesoflavibacter, suggests potential roles in biofilm formation and host-microbe interactions.36
The most prominent family of bacteria in the phylum Bacteroidetes is the Flavobacteriaceae, which includes rod-shaped, Gram-negative bacteria that do not produce spores. These bacteria are chemoorganotrophic, primarily relying on respiratory metabolism, and thrive in freshwater, marine, and terrestrial environments.37 In aquatic ecosystems, they are often associated with surfaces such as algae, fish, or organic detritus.38 Members of this family have also been noted for producing bioactive compounds with antirachitic, cell growth-promoting, and antioxidative properties.37 Their prevalence in this study suggests their ecological importance in the marine habitats occupied by Hippocampus barbouri. Bacillaceae, another significant family identified, is characterized by its ability to form endospores, which confer resistance to environmental stresses such as heat, radiation, and chemicals. These bacteria are ubiquitous in fresh and marine waters and are also present in lumens of humans, animals, and even in fermented foods.39 Members of Bacillaceae are known for synthesizing antibiotics and other bioactive proteins, highlighting their potential role in maintaining the microbial balance on seahorse skin while contributing to the environmental microbial diversity.40 Pseudomonadaceae is a family with members commonly found in soil, water, and skin flora, as well as in artificial environments. The presence of Pseudomonadaceae on seahorse skin aligns with its broad adaptability and potential role in maintaining skin health and microbial balance. Moraxellaceae includes bacteria frequently associated with terrestrial and marine animals, including fish and crustaceans. These bacteria are often considered part of the normal flora and have been isolated from diverse environments such as deep-seawater surfaces, sea ice, and soil.41,42 Members of this family are known for their role in bioremediation and antibacterial properties,43 suggesting their potential ecological role in marine environments. Another prominent family present in this study is Vibrionaceae. Members of this family are abundant in aquatic environments, inhabiting water columns, plankton, and marine animal tissues.44 While some species are pathogenic, others have demonstrated important ecological and medical roles, such as their fermentative metabolism and potential for biotechnological applications.45 Their presence reflects the environmental interactions of H. barbouri in its habitat. Aeromonadaceae, commonly found in aquatic ecosystems, including freshwater, seawater, and sediments, was also identified. Although their potential applications in biotechnology and medicine are less explored, they are known to impact host health, as significant doses have been linked to self-limiting diarrhea.46 The bacterial families observed in this study reflect the environmental conditions and ecological interactions of H. barbouri. Their presence highlights the complex microbial dynamics associated with marine organisms and underscores their potential for biotechnological and ecological applications.
Overall, 61 unique bacterial species were identified, with 80% shared between both male and female. The majority of these species belong to Pseudomonadota (formerly Proteobacteria). Members under Pseudomonadota are often associated with critical roles in nutrient cycling, organic matter degradation, and symbiotic relationships with marine organisms. Their abundance in aquatic environments further supports their adaptability to dynamic conditions, including variations in salinity, temperature, and nutrient availability,47 which makes them essential for maintaining health of the marine environment and habitants including H. barbouri. The shared presence of these bacterial communities likely reflects a symbiotic relationship between the bacteria and their host, providing mutual metabolic benefits.48 The abundance of Pseudomonadota in this study suggests that H. barbouri harbors a rich and diverse bacterial community on its skin, which is consistent with the observation that the balance between the number of bacteria adhering to the skin and the host’s ability to support them as they are vital in determining the normal skin microflora for a given species.49
The successful identification of bacteria present in the epidermal surface of male and female Hippocampus barbouri underscores the efficiency of utilizing 16S rRNA amplicon sequencing analysis. The bacterial composition reveals notable differences in species richness between males and females, with several factors influencing these variations in diversity. These factors include sex, species interactions, size of home ranges, immobility rates, and habitat disturbances. Building on previous research on microbial communities, this study offers a detailed assessment of the abundance and variety of microbial species associated with H. barbouri. Furthermore, as baseline data, it serves as a valuable reference for future studies aimed at further characterizing the microbial communities of this species.
ACKNOWLEDGMENTS
The authors gratefully acknowledge the funding support provided by DOST-ASTHRDP. Sincere appreciation is also extended to the Bureau of Fisheries and Aquatic Resources (BFAR), the local government unit of Surigao del Norte, and the officials and residents of Cantiasay and Hanigad Islands, as well as the Mordeno and Adalla families for their invaluable assistance and cooperation.
CONFLICT OF INTEREST
The authors declared that there is no conflict of interest.
AUTHORS’ CONTRIBUTION
RCMHOK and SRMT conceptualized the study. RCMHOK and CSPA applied methodology. CMHOK and CSPA performed investigation and visualization. RCMHOK and SRMT performed formal analysis and funding acquisition. SRMT performed supervision. RCMHOK and SRMT wrote and edited the manuscript. All authors read and approved the final manuscript for publication.
FUNDING
This study was funded by DOST-ASTHRDP to RCMHOK through a scholarship grant.
DATA AVAILABILITY
The datasets generated and/or analysed during the current study are available in the GenBank database at NCBI vide BioProject PRJNA1217945, accession number SRR32211362.
ETHICS STATEMENT
This study was approved by the Bureau of Fisheries and Aquatic Resources (Department of Agriculture, Republic of the Philippines, Quezon City) under Gratuitous Permit No. 0249-23.
- Oconer E, Herrera A, Amparado E, Dela Paz R, Kime D. Reproductive morphology and gonad development of the male seahorse, Hippocampus barbouri Jordan and Richardson 1908. Asia Life Sci. 2003;12(1):27-38.
- Szekely T. Evolution of reproductive strategies: sex roles, sex ratios and phylogenies. Biol Futura. 2023;74(4):351-357.
Crossref - Sehnal L, Brammer-Robbins E, Wormington AM, et al. Microbiome composition and function in aquatic vertebrates: small organisms making big impacts on aquatic animal health. Front Microbiol. 2021;12:567408.
Crossref - Ortega RCMH, Tabugo SRM, Martinez JGT, Padasas CS, Balcazar JL. Occurrence of Aeromonas species in the cutaneous mucus of Barbour’s seahorses (Hippocampus barbouri) as revealed by high-throughput sequencing. Animals. 2023;13(7):1241.
Crossref - Bereiter-Hahn J, Richard KS, Elsner L, Voth M. Composition and formation of flame cell caps: a substratum for the attachment of microorganisms to seahorse epidermis. Proc R Soc Edinb B Biol Sci. 1980;79(1-3):105-112.
Crossref - Bennice CO, Krausfeldt LE, Brooks WR, Lopez JV. Unique skin microbiome: insights to understanding bacterial symbionts in octopuses. Front Mar Sci. 2024;11:1448199.
Crossref - Lema NK, Gemeda MT, Woldesemayat AA. Recent advances in metagenomic approaches, applications, and challenges. Curr Microbiol. 2023;80(11):347.
Crossref - Vincent ACJ, Evans KL, Marsden AD. Home range behaviour of the monogamous Australian seahorse, Hippocampus whitei. Environ Biol Fishes. 2005;72(1):1-12.
Crossref - Gomez JA, Primm TP. A slimy business: the future of fish skin microbiome studies. Microb Ecol. 2021;82(2):275-287.
Crossref - Ortega RCMH, Tabugo SRM, Martinez JGT, Padasas CS, Balolong MP, Balcazar JL. High-throughput sequencing-based analysis of bacterial communities associated with Barbour’s seahorses (Hippocampus barbouri) from Surigao del Norte, Philippines. Lett Appl Microbiol. 2021;73(3):280-285.
Crossref - Balcazar JL, Loureiro S, Da Silva YJ, Pintado J, Planas M. Identification and characterization of bacteria with antibacterial activities isolated from seahorses (Hippocampus guttulatus). J Antibiot. 2010;63(5):271-274.
Crossref - Quast C, Pruesse E, Yilmaz P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41(D1):D590-D596.
Crossref - Pruesse E, Peplies J, Glockner FO. SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics. 2012;28(14):1823-1829.
Crossref - Li W, Godzik A. CD-HIT: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22(13):1658-1659.
Crossref - Camacho C, Coulouris G, Avagyan V, et al. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10(1):421.
Crossref - Ondov BD, Bergman NH, Phillippy AM. Interactive metagenomic visualization in a web browser. BMC Bioinformatics. 2011;12(1):385.
Crossref - Felsenstein L. Confidence limits on phylogenetics: an approach using the bootstrap. Evolution. 1985;39(4):783-791.
Crossref - Kumar S, Stecher G, Tamura K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger dataset. Mol Biol Evol. 2016;33(7):1870-1874.
Crossref - Wakeley J. Substitution-rate variation among sites and the estimation of transition bias. Mol Biol Evol. 1994;11(3):436-442.
Crossref - Garlet A, Andre-Frei V, Del Bene N, et al. Facial skin microbiome composition and functional shift with aging. Microorganisms. 2024;12(5):1021.
Crossref - Bullington LS, Lekberg Y, Larkin BG. Insufficient sampling constrains our characterization of plant microbiomes. Sci Rep. 2021;11(1):3645.
Crossref - Vincent ACJ. The international trade in seahorses. Cambridge, UK: TRAFFIC International. 1996.
- Consi TR, Seifert PA, Triantafyllou MS, Edelman EA. The dorsal fin engine of the seahorse (Hippocampus sp.). J Morphol. 2001;248(1):80-97.
Crossref - Warfe DM, Barmuta LA. Habitat structural complexity mediates the foraging success of multiple predator species. Behav Ecol. 2004;141(1):171-178.
Crossref - Garrick-Maidment N, Jones L. British seahorse survey report 2004. Devon, England: The Seahorse Trust. 2004.
- Lamb SF. Investigating differing aquaria environments and their influence on natural behaviours and breeding patterns of captive-bred short-snouted seahorse, Hippocampus hippocampus (Linnaeus, 1758). Thesis No. 10217865 2013.
- Gristina M, Pierri C, Lazic T, Palma J. Behavioral traits of captive short-snouted seahorse Hippocampus hippocampus, Linnaeus 1758. IEEE Conference Publication. 2022.
Crossref - Caldwell I, Vincent ACJ. A sedentary fish on the move: effects of displacement on long-snouted seahorse (Hippocampus guttulatus Cuvier) movement and habitat use. Environ Biol Fishes. 2012;96(1):67-75.
Crossref - Kloska A, Cech GM, Sadowska M, Krause K, Szalewska-Palasz A, Olszewski P. Adaptation of the marine bacterium Shewanella baltica to low-temperature stress. Int J Mol Sci. 2020;21(12):4338.
Crossref - Zrneic S. Microbial diseases of marine organisms. J Mar Sci Eng. 2022;10(11):1682.
Crossref - Zhao Y, Wei HM, Yuan JL, Xu L, Sun JQ. A comprehensive genomic analysis provides insights on the high environmental adaptability of Acinetobacter strains. Front Microbiol. 2023;14:1177951.
Crossref - Ghose M, Parab AS, Manohar CS, Mohanan D, Toraskar A. Unraveling the role of bacterial communities in mangrove habitats under the urban influence using a next-generation sequencing approach. J Sea Res. 2024;198:102469.
Crossref - Pohlner M, Dlugosch L, Wemheuer B, Mills H, Engelen B, Reese BK. The majority of active Rhodobacteraceae in marine sediments belong to uncultured genera: a molecular approach to link their distribution to environmental conditions. Front Microbiol. 2019;10:659.
Crossref - Alattas H, Glick BR, Murphy DV, Scott C. Harnessing Pseudomonas spp. for sustainable plant crop protection. Front Microbiol. 2024;15(24):1485197.
Crossref - Marques PH, Prado LCDS, Felice AG, et al. Insights into the Vibrio genus: a One Health perspective from host adaptability and antibiotic resistance to in silico identification of drug targets. Antibiotics (Basel). 2022;11(10):1399.
Crossref - Lin Y, Briandet R, Kovacs AT. Bacillus cereus sensu lato biofilm formation and its ecological importance. Biofilm. 2022;4:100070.
Crossref - McBride MJ. The family Flavobacteriaceae. In: Rosenberg E, DeLong EF, Lory S, Stackebrandt E, Thompson F, eds. The Prokaryotes. Springer, Berlin, Heidelberg. 2014:643-676.
Crossref - Gavriilidou A, Gutleben J, Versluis D, et al. Comparative genomic analysis of Flavobacteriaceae: insights into carbohydrate metabolism, gliding motility, and secondary metabolite biosynthesis. BMC Genomics. 2020;21(1):569.
Crossref - Sivamaruthi BS, Alagarsamy K, Suganthy N, Thangaleela S, Kesika P, Chaiyasut C. The role and significance of Bacillus and Lactobacillus species in Thai fermented foods. Fermentation. 2022;8(11):635.
Crossref - Crivelli XB, Cundon C, Bonino MP, Sanin MS, Bentancor A. The complex and changing genus Bacillus: a diverse bacterial powerhouse for many applications. Bacteria. 2024;3(3):256-270.
Crossref - Teixeira LM, Merquior VLC. The family Moraxellaceae. In: Rosenberg E, DeLong EF, Lory S, Stackebrandt E, Thompson F, editors. The Prokaryotes. Springer, Berlin, Heidelberg. 2014.
Crossref - Golas I, Szmyt M, Potorski J, Lopata M, Gotkowska-Plachta A, Glinska-Lewczuk K. Distribution of Pseudomonas fluorescens and Aeromonas hydrophila bacteria in a recirculating aquaculture system during farming of European grayling (Thymallus thymallus L.) broodstock. Water. 2019;11(2):376.
Crossref - Yao K, Liu D. Moraxella catarrhalis. Mol Med Microbiol. 2024:1503-1517.
Crossref - Kokashvili T, Whitehouse CA, Tskhvediani A, et al. Occurrence and diversity of clinically important Vibrio species in the aquatic environment of Georgia. Front Public Health. 2015;3:232.
Crossref - Sampaio A, Silva V, Poeta P, Aonofriesei F. Vibrio spp.: life strategies, ecology, and risks in a changing environment. Diversity. 2022;14(2):97.
Crossref - Greiner M, Anagnostopoulos A, Pohl D, Zbinden R, Zbinden A. A rare case of severe gastroenteritis caused by Aeromonas hydrophila after colectomy in a patient with anti-Hu syndrome: a case report. BMC Infect Dis. 2021;21(1):1097.
Crossref - Suominen S, Dombrowski N, Sinninghe Damste JS, Villanueva L. A diverse uncultivated microbial community is responsible for organic matter degradation in the Black Sea sulphidic zone. Environ Microbiol. 2021;23(6):2709-2728.
Crossref - Li Y, Huang Y, Wronski T, Huang M. Diversity of bacteria associated with lichens in Mt. Yunmeng in Beijing, China. PeerJ. 2023;11:e16442.
Crossref - Pardo A, Villasante A, Romero J. Skin microbiota of salmonids: a procedure to examine active bacterial populations using an RNA-based approach. Appl Microbiol. 2023;3(2):485-492.
Crossref
© The Author(s) 2025. Open Access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License which permits unrestricted use, sharing, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.