


ISSN: 0973-7510
E-ISSN: 2581-690X
Shubhangi Rautela1 and Monika Singh1
Global population is increasing exponentially, besides this, the number of oncogenic patients also increases globally. Among the all types of cancers, stomach cancer patients make a huge number, worldwide. In gastrointestinal oncology, some urease producing microbes are the core cause of adenocarcinoma. One of the most prominent bacteria is Helicobacter pylori, which is a flagellated, microaerophilic proteobacteria that adheres in the stomach epithelial cells. Among the half of the human population of the world, which are suffering from gastric ulcers, cancer and number of genetic disorder. In this review, authors have summarized the pathophysiology and molecular mechanism of the H. pylori infection in human being throughout the past ten years. H. pylori, expresses various virulence factors, and display a variety of adaptive mechanism during colonization and adhesion in the gastric region. This bacterium also produces several cytotoxins to speed up effective colonization in the host. Nonetheless, several number of techniques have been developed to identify the virulence genes of H. pylori infection. Furthermore, alternative treatment approaches are frequently using to eradicate the disease such as antibiotics and plant-based medicines. Currently the prescribed course of treatment for H. pylori combines with antimicrobial drugs like amoxicillin, clarithromycin, metronidazole, and levofloxacin, but now days these medicines are less effective against this bacterium, data were obtained when discuss with the experienced Gastroenterologist. At present, various research studies are being conducted to create effective vaccinations to fight the H. pylori infection; it has additionally been a goal of several running research projects. This review article might be helpful for the researchers who wish to work on novel drug designing, novel identification and treatment methods of H. pylori which is a necessity of gastrointestinal oncology.
Helicobacter pylori, Virulence, Gastric Cancer, Antibiotic Resistance, Alternative Therapy
Microbes are universal; they are diverse from human body to forest and ocean. The (GI) gastrointestinal tract of human being is a harbour of millions of microorganisms; some are beneficial some are pathogenic. The pathogenic microbes interact with the host to affect human health.1 Among the pathogenic microbes, Helicobacter pylori is the most studied microbial pathogen in human gastrointestinal tract, it appears to have started in East Africa and spread about 58,000 years ago.2 Several studies revealed that, it is linked to the number of gastrointestinal abnormalities, including stomach ulcers, peptic ulcers, and stomach cancer also and result in severe upper gastrointestinal conditions such chronic gastritis, dyspepsia. Furthermore, a variety of other conditions, including as autoimmune diseases, cardiovascular and cerebrovascular problems, and iron-deficiency anaemia, have been shown to be associated with H. pylori infection.3-6 At global scale, approximately 70% of stomach cancers is caused due to H. pylori infection.7 According to epidemiology, about 30%-50% and 50%-60% population having H. pylori infections in developed and developing countries respectively.8
Many infections start at infancy, when recipients are healthy individuals who subsequently develop symptoms as adults. But the vast majority of infected people lack symptoms of H. pylori infection.9 This infection is contagious and spread by different pathways, including oral-oral, fecal-oral, and environmental transmission via polluted water sources.10-12 Besides these, some environmental variables, including food, carcinogens exposure, excessive alcohol use and smoking are important causes of H. pylori infection.13 In addition, the persistence of this bacterium in the host is greatly influenced by class switching. This pathogen can survive in the digestive system of the host by shape switching such as spiral to coccoid shape.14 H. pylori uses flagella-mediated motility to move towards the stomach’s epithelial cells and damage the mucous layer.15 The H. pylori bacteria that grows as cell-associated micro-colonies in the stomach after entering the stomach cell-lines. In addition, the bacterial movement is a crucial mechanism for effective colonization, establishment and infection in human gut. Moreover, chemotaxis allows a bacterium to change, how it swims in response to chemical signals from the extracellular environment.16 Pathogenicity of H. pylori consist of indirect inflammatory reactions and host signaling pathways inside the stomach mucosa, whereas antibiotic resistance strategies used by the bacteria include genetic mutations and biofilm formation.17,18 Furthermore, H. pylori pathogenicity depends on the strain, even some other factors are also involved. The interaction between the host and the bacterium is facilitated by the expression of a particular virulence factors.19 A number of factors influence the rate at which an H. pylori infection spreads, such as the virulence of the strain, the efficacy and genetic composition of the host’s immune system, the patient’s dietary habits, and medical history.20
Among the all diagnostics methods, two specific category of diagnostics methods are using frequently; one is invasive another is non-invasive. Invasive diagnostics methods includes endoscopic examination, histopathology, rapid urease test (RUT). Whereas Urea breath test, stool antigen test, serology, and molecular diagnostic techniques such as polymerase chain reaction are examples of non-invasive techniques.21 Although a serologic test for antibodies can detect bacterial exposure, it cannot detect ongoing infection. Accessibility, cost, clinical circumstances, any gastric cancer family history, and pre-test likelihood of infection, which may be affected by population frequency of infection, these all plays an important role in the selection of an appropriate test. The recommended course of treatment for H. pylori illness is triple antibiotic therapy (sometimes known as quadruple therapy). Different medication combinations, sequential treatments, and concurrent therapies have also been employed as effective substitutes for H. pylori treatment.
In this review article, the authors are trying to compile the current knowledge about the methods and mechanism of colonization of H. pylori in the gastrointestinal region and how it affects human health and useful internal microbiota. Nowadays, the elimination of H. pylori has become more challenging due to antibiotic resistance development in the strain. It will serve to highlight the most recent information about the mechanism of disease and virulence of H. pylori infection, it might be helpful for the core researchers of this field.
Molecular mechanism of infection and virulence of H. pylori
The genetically diversified bacteria H. pylori have been co-evolving with humans for more than 60,000 years ago. Because of its genetic flexibility, it has generated a persistent infection that is connected to gastrointestinal disorders. This long-standing connection has resulted in the development of host-specific adaptive changes, which have created unique genotypes. It has been demonstrated that different genotypes are present and they are more prevalent in different geographical areas. The severity of gastrointestinal disorders linked to H. pylori has been associated with the predominant genotype in each area.22 Warren and Marshall was the eminent microbiologist who originally identified H. pylori.23 Moreover, in the year 1997, the entire genome of H. pylori was sequenced, it was a great achievement to know the molecular mechanism of virulence of H. pylori in human.24,25 Since then, many research article have been published on H. pylori’s pathology, pathogenicity, immunology, and pathogenesis. However, geographically, H. pylori infection varies, with developing countries having the highest burden of infection.26 It has the ability to colonize a host, reproduce, and endure.13 In the pathogenesis process, when the pathogen reaches into the stomach, they use their urease activity to combat the unfavourable acidic environment. The cell then uses flagella-mediated motility to travel in the direction of the stomach epithelium. Effective colonization and ongoing infection are caused by additional interactions between H. pylori adhesins and the host cell’s receptors. It creates a range of toxins and chemicals that causes harm to the host tissues adhering to successful colonization. However, chemical messengers that are released during an infection is triggered innate immunity. Additionally, neutrophil activation and associated clinical manifestation are present.27 Besides this, biofilms also produced by H. pylori to ease the attachment infection, colonization and persistence. By reducing its resistance to antibiotics, the biofilm helps to cause mutations that make bacterial elimination more challenging.28 This strategy increases genetic material interchange and encourages recombination more often.29 Furthermore, it has been observed that the growth of biofilms is positively associated with an increase in the expression of several genes those are important for virulence and resistance to antibiotics.30,31 Common symptoms of the infection includes nausea, vomiting, burping, and loss of appetite. Additional signs include in some cases are, abdominal cramps, weight loss and indigestion.32
There are several ways to identify H. pylori infection, such as invasive & non-invasive methods, which are discussed in detail (Figure 1).21 Every technique has advantages and disadvantages. Moreover, Proton pump inhibitors, antibiotics, and medicines containing bismuth are additional variables that may have an impact on how accurate particular results from tests remain. Additionally, H. pylori and the virulence gene are found using real-time PCR methods.33 To figure out H. pylori virulence genes and its variants, the polymerase chain reaction (PCR) is frequently used, however it can be time-consuming and expensive.34 Triple antibiotic therapy (quadruple technique) is one of the most standard treatments of the H. pylori infection. In this, alternative treatments like phytomedicines (plant-based medicines) and antibiotics have been used to increase eradication rate of infection, the current course of treatment still combines antimicrobial drugs like amoxicillin, clarithromycin, metronidazole and levofloxacin with anti-secretory drugs like proton pump inhibitors (PPIs).35,36
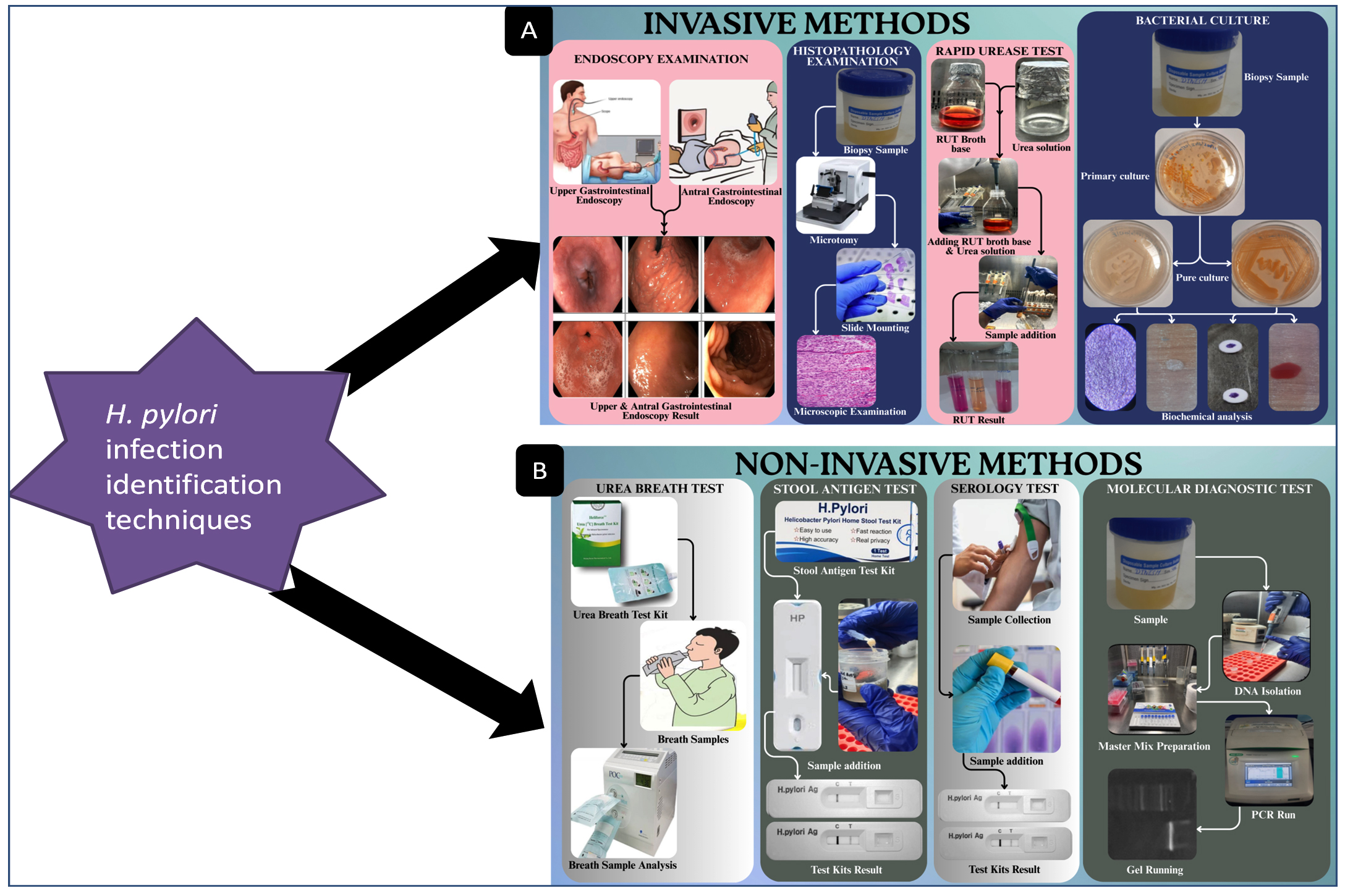
Figure 1. Graphical presentation of H. pylori infection identification techniques: (A): Invasive methods, (B): Non-invasive methods
Defense mechanism
Epithelial cell: the initial line of protection
The human gastrointestinal region’s first-line defensive barrier, or epithelial cell, works by forming a tight structure which prevents pathogen adhesion, growth, and movement. Pathogens like H. pylori employ their specific mechanisms to adapt to the hardest lumen conditions and colonies the host, and then start to spread infection. H. pylori colonization rely on four main processes: adaptation to the stomach mucosa’s acidic environment; migration and penetration towards the primary defense barrier (epithelial cell) through flagella-mediated movement; attachment to a particular receptor cell by adhesins molecules, and the last one by damaging to the tissue cell and other adverse effects by released toxin. After a severe H. pylori infection, the bacterium can adhere to a host cell and survive in the acidic environment of the stomach, and also release cytotoxic proteins such as, blood group antigen-binding adhesion (BabA); outer inflammatory protein (OipA); outer membrane protein (OMP); outer membrane vesicles (OMV); vacuolating cytotoxins (VacA) high-temperature requirement A (HtrA), neutrophil-activating protein A (NepA), and sialic acid binding adhesins (SabA). It also has the outer membrane proteins (OMPs) such as HopQ and HopZ and some other proteins i.e HomA, HomB, HomC, and HomD of H. pylori. According to recent research study by Xu et al., concluded that HomB protein can cause IL-8 secretion which plays a significant role in cancer development and progression. Lipoproteins A and B (AlpA/AlpB) also assist H. pylori in colonization, attachment to epithelial cells, and even the formation of biofilm.37 Other weakly adhering cytotoxins also have a significant role in H. pylori infection in human (Figure 2).
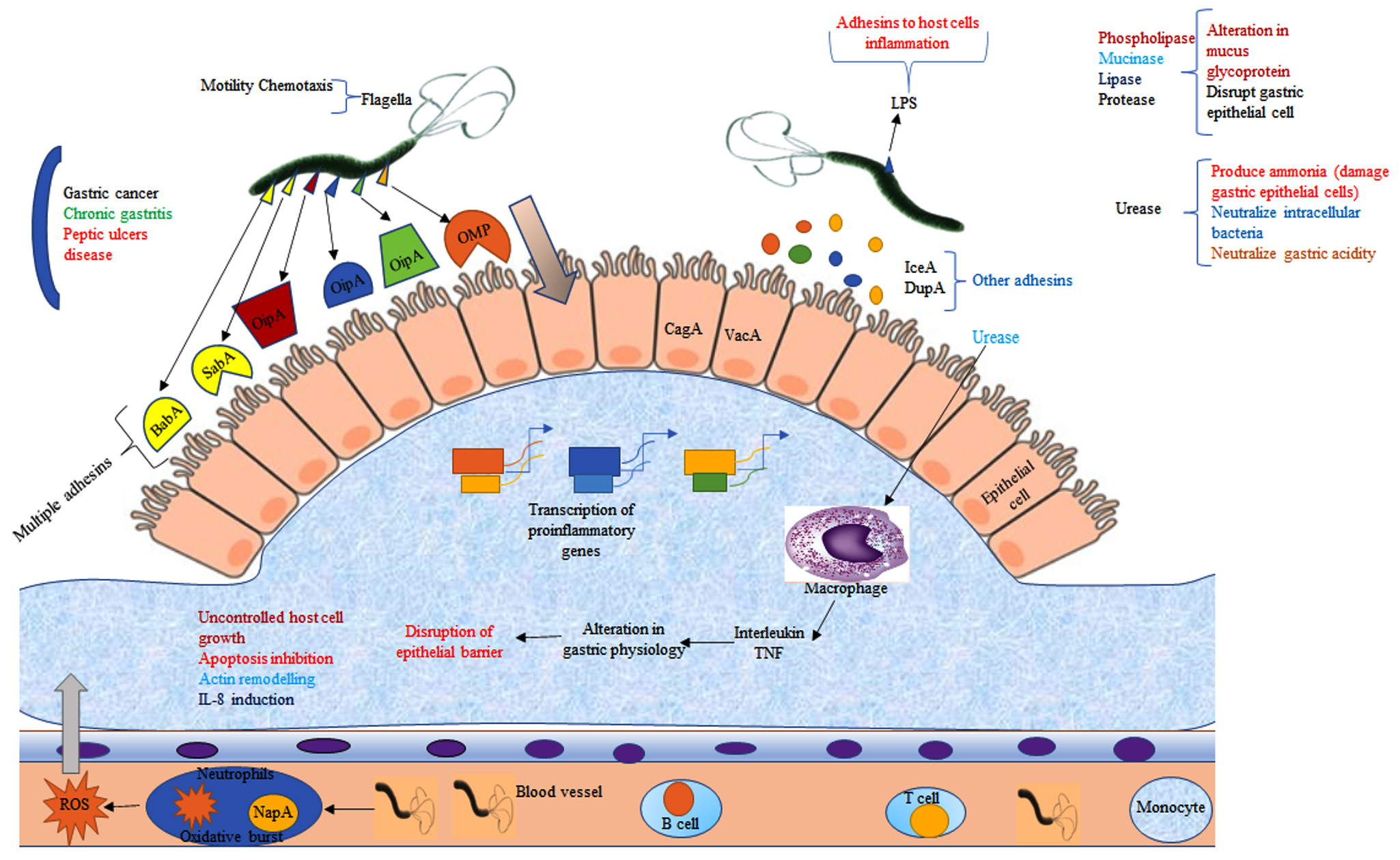
Figure 2. Graphical illustration of pathophysiology and pathogenicity of H. pylori infection and disease establishment via colonization
Urease production and adaptation to the stomach mucosa’s acidic environment
In order to function physiologically and as cofactors for enzyme reactions for the adaptation to the stomach mucosa’s acidic environment, several transition metals are essential for the organism.38 Certain metals are essential to the survival and growth of microorganisms, such as nickel, which is a cofactor for the enzyme’s urease and hydrogenase, 24 nickel ions were needed to catalyze one active urease molecule.39,40 In this context, the bacterial virulence proteins FecA3 and Frp B4, which are present on the outer membrane of the bacterial cell wall, these proteins are frequently used for the acquisition of metal ions, which are found in blood.41 Through the protein channel NixA, nickel ions are moved from its outer membrane into its inner cytoplasmic membrane of the bacterium.42 For several bacterial species, urease has been discovered to be a key enzyme for the pathogenicity via colonization in the gastric mucosa.43 It can hydrolyze the urea into ammonia and CO2 which can help to reduce acidic environment and facilitate colonization.38 H. pylori often used molecular hydrogen as a fuel source for its metabolic process because the hydrogenase’s participation in an alternate signaling route.44 However, urease cannot be triggered in the presence of inadequate amount of nickel molecules. Therefore, a significant amount of nickel ion is needed for its metabolic process, which might lead to cell death. In order to minimize the effects of the harsh environment in the gastric region, the cluster of urease gene in H. pylori bacteria trigger the synthesis of the urease enzyme. These urease genes includes catalytic units (urea A&B), acid-gated urea channel (ureI), and auxiliary assembly proteins (ure E-H), these are produces in the organism.37 Besides this, the pH of the gastrointestinal regions affects urea production. When the gastrointestinal tract’s pH is neutral (7.0), then the urea channel is blocked, and it opens when the environment turns to acidic, Low pH levels may inhibit the synthesis of proteins, urease, and catalase function.17 The primary process involved in the synthesis of urease is the transformation of shape of H. pylori from spiral to coccoid. The spiral shaped bacterial strains has more urease activity than the coccoid forms.45
Shape Switching of H. pylori
Shape switching is key character of H. pylori. When unfavorable environmental conditions comes, bacteria may change their morphology from spiral to coccoid. The bacterium’s coccoid form may remain for a long time, is easily non-culturable, antibiotic-resistant, capable for replication, and can create a number of bad effects to the stomach mucosa. The bacteria’s coccoid viable not-culturable form represented as ‘CVNCF’. Whereas the bacterial spiral shape forms are culturable. They are representing as spiral viable culturable form ‘SVCF’ in the entire manuscript. Moreover direct culture techniques are ineffective to detect the coccoid forms of the bacteria; to overcome the problem, molecular methods and direct electron microscopy can be used. H. pylori are able to survive by changing its shape through the SVCF to the CVNCF during the most unfavourable environmental conditions such as lack of nutrients, absence of oxygen, desiccation, exposure to antimicrobial agents, and the stomach’s acidic fluid. The CVNCF bacterium nevertheless maintains their pathogenicity and metabolic activity, and they are capable of performing this while they are in a culturable and viable state.46 The Elhariri et al. research revealed that H. pylori may survive in unfavorable conditions. Through the use of nested PCR, approximately 75 samples of H. pylori were isolated for the research purposes from animal faecal matter and milk. There was a total of 13 samples that were containing the spiral forms (SVCF) of H. pylori, whereas the other samples could not be cultured, suggesting that the presence of the CVNCF form of the bacteria. The SVCF was cultivated in milk for 1, 5 and 10 days at 4 °C, 37 °C and 40 °C, respectively, to illustrate this study’s attempt to demonstrate the survivability under challenging environmental conditions. A microbial load reduction also observed between one and ten days. It was found that the CVNCF forms, which can survive in UTH milk for thirty days or more, appeared at the same time as SVCF began to decline. The genotype of both cultures (SVCF and CVNCF) is identical in the infected individual, which indicated that the spiral form (SVCF) has evolved into the non-culturable form (CVNCF)46 (Figure 3).
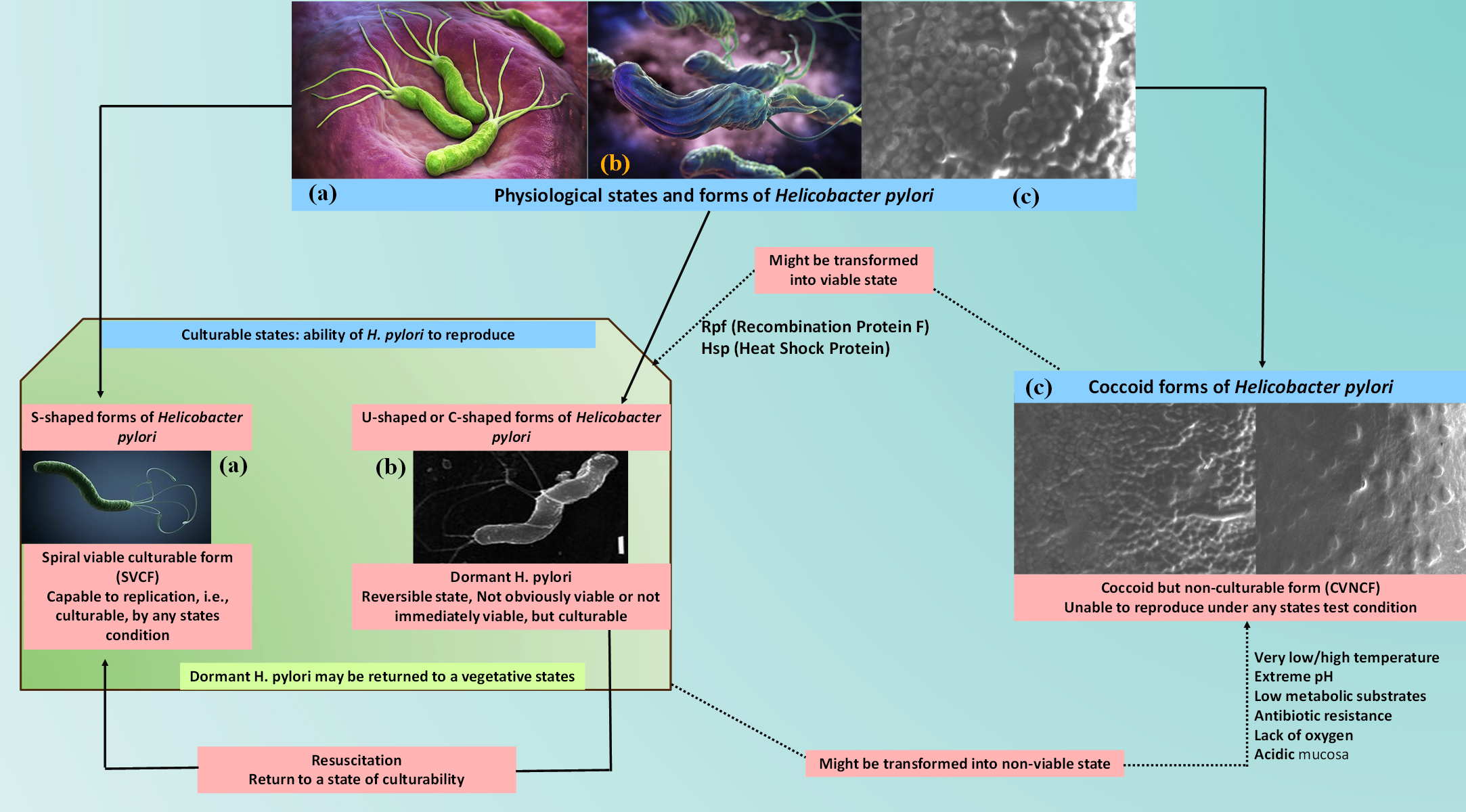
Figure 3. Graphical illustrations of shape switching of H. pylori
Migration and penetration of the epithelial cell
The H. pylori are flagellated and gram-negative bacteria. They pass through the epithelial mucus membrane to the underlying membrane under the neutral pH condition. According to some research studies, in which the bacterial flagella are more important to migration, penetration, colonization, and infection in the gastric mucosa. Actually, the bacterial flagella, which have three structural components (basal body, hook, and filament), are a complicated organelle of the bacterium. In the structural component of flagella, FlaA and FlaB proteins play a crucial role in making up the filaments. FlaB is located in the flagellum’s base, whereas FlaA is located in the outer area. The flagella can be observed when a pathogen is swimming, swarming, or spreading. H. pylori move and maintain adhesions on the epithelial cell via its flagella. Moreover the stomach high acidic environment facilitate flagella to move more quickly.32 At this acidic pH, a proton motive force drives the proteins which control the flagellar movement. When mutations present in the flagellar structural genes such as FliD, FlaA, or FlaB, can prevent the H. pylori from colonizing the stomach mucosa. In a study, the result concluded that FlaB and FlaA deficient strains of H. pylori are completely incapable to synthesize the flagella. Moreover, a FlaB defective bacterial strain have decreased motility and adhesion abilities.27 Result concluded that, a non-flagellated bacterium does not colonize the mucosal epithelial cell.29
Attachment to a particular receptor cell by adhesins molecules
H. pylori must be adhered to the epithelial cell after entering the stomach mucosa in order to initiate infection. The gastric mucosa’s surface possesses receptors for H. pylori adhesins that bind to the mucus line. Although the two adhesins proteins that received the most attention are sialic acid-binding adhesin (SabA) and blood-antigen binding protein A (BabA), which are absent in certain H. pylori strains. Besides these two adhesions, Neutrophil-activating protein (NAP), heat shock protein 60 (Hsp60), adherence-associated proteins (AlpA and AlpB), H. pylori outer membrane protein (HopZ), and lac di NAc-binding adhesin (LabA) are a few other adhesins help in adapting and infection of H. pylori in the hosts. Moreover, these proteins also provide the bacterial nutrition, promotes the release of bacterial cytotoxins, and produces other harmful chemicals in the host cell.37
Neutrophil activated protein A
H. pylori secrete Neutrophil activated protein (NAP), it has the property of DNA-protecting proteins under the unfavorable environment, which significant similar structure with iron storing proteins. NAP was initially discovered to produce large amount of oxygen radicals from neutrophils, which damages nearby cells and increases neutrophil adhesins to endothelium cells during bacterial infection.47 The acquisition of β2 integrin surface receptors on the neutrophil surface membrane is necessary for this NAP-induced adhesion. Moreover, NAP also triggers the production and release of IL-8, macrophage inflammatory protein by neutrophils. Thus, NAP is tightly linked with the characteristic of chronic gastritis and the passage of neutrophils and mononuclear cells into the stomach mucosa, both of them are caused by bacterial infections. In the context of molecular mechanism, when neutrophils come to contact with the NAP on the surface of bacteria, their glycosphingolipids function act as a receptor.48 Furthermore, NAP is thought to help to Sialic acid-binding adhesion on the outer coating of the host cell.49 Several research studies revealed that NAP can prevent to form free radicals from destroying H. pylori DNA by adhering to it.50 It’s interesting to note that NAP can cause T helper cells to develop into T helper 1 phenotypes and encourage neutrophils or monocytes to boost IL-12 production.51 Therefore, NAP has been proposed as an adjuvant for vaccination and an immunotherapeutic anticancer drug in clinical applications. However, additional research is needed to verify that activation of glucose analogue (2-Deoxy-D-Glucose) by NAP can be treatment of DG-based cancer immunotherapy and cancer vaccines.
BabA and BabB: Blood group antigen-binding proteins
There are three different kinds of blood group antigen-binding adhesin known as babA1, babA2, and babB. The babA protein is encoded by babA2. Despite having very similar coding sequences, babA1 does not have the translation start codon. H. pylori uses BabA to bind to the fucosylated Lewis B blood-group antigen (Lewis’s b [Leb]) that is expressed on the cells of the host gastric epithelium during the initial infection of the human stomach.52 The BabA receptor shares structural similarities with the O type blood antigen, and epidemiological data shows a link between the blood type O and illnesses of the stomach.53 The majority of the sequence variation between blood-antigen binding protein babA and babB occurs in their central sections, which are located in their 5′ and 3′ regions, respectively. The central portion of the babA sequence, which is essential controls babA for adhesion. In western nations, babA expression has been connected to a higher risk of stomach cancer. Nevertheless, among the Asians, the presence of babA is not associated with gastric-related disorders.54 babB’s purpose is still unknown, however patient expression of babB was linked to more stomach abnormalities.55
However, certain H. pylori strains have a chimeric babB/A that can attach to the Leb antigen through genetic recombination of babA1 and babB under specific conditions, even though they lack babA2 in their genome or have a mutation that causes a deficiency in BabA.56 One research study found that H. pylori bacterium was isolated from the samples of chronically infected people, to analyze and learn about the dynamics of Leb adhesion all over human infection. The results revealed that the BabA-Leb binding phenotype is remarkably durable during long-term human infection.56,57
Sialic acid-binding adhesin (SabA)
The occurrence of sialyl-Lewis x glycosphingolipid (sLex) antigen increase on the cell’s surface at sites of severe local inflammation caused by H. pylori. This indicates that SabA adhesin is essential for helping H. pylori attachment and colonization of gastric epithelial cells in a gastritis patient.58 Lex and Ley antigens are frequently encountered in the LPS of H. pylori strains. These two Lex and Ley antigens were strongly associated with H. pylori colonization.59 Although the sabB and sabA genes are similar.60 Therefore, it is important to do more research on how sabB affects bacterial adherence and pathogenicity. Nearly 80% of clinical strains include the sabA gene, and the sequencing data showed two different sabA genotypes. In the molecular sequencing, among the nucleotides, CT (cytosine and Thymine) repeat several sequences repeat CT, which are located in the 5′ ORF of sabA controls the synthesis of type I sabA, which can be controlled by slipped strands mispairing (SSM). Only strains with 4, 7, or 10 CT dinucleotide repeats and an ORF allowing for the entire length of SabA are able to synthesize the functional protein.58 The status of this expression is “On”. Once the CT repeat number is adjusted to 3, 5, 6, 8, 9, and 11, the frame-shift will be producing early stop codons in the ORF. In this state, SabA is expressed in a shortened form and the gene expression is “Off”. The elimination of the CT repeated and the distinct sequence in this area are specific to the type II sabA. Additionally, the 5’ coding areas of not only the sabA gene but also the genes for the adhesins oipA and hopZ have been used to identify by CT dinucleotide repeats.61
The western blotting-detected expression of SabA differs significantly to the sequence-based assumption.58 In type I strains, which should be “On,” Western blotting only finds nearly 43% expression of SabA. Kao et al. stated that SSM also causes a change in the length of a poly T tract in the sabA promoter region.62 Furthermore, it is believed that the poly T tract’s length affects the activity of the sab A promoter, providing an alternative means of regulating transcription in H. pylori, which has few conventional trans-acting regulatory factors.62 During H. pylori infection, SSM create a mixed-genotype population that may help the bacteria evade the immune system and adapting to different hosts. Analysing clinical data reveals that the population of H. pylori in the body is significantly higher in patients infected with SabA-positive strains than those patients infected with SabA-negative strains. These studies indicated that the patients which have low or absent Leb expression, SabA’s interaction with the sLex antigen may promote H. pylori colonization.58
Even though the main host factor controlling H. pylori density in patients with a babA2-positive H. pylori infection is Leb level, H. pylori may benefit from the deficiencies of sLex-mediated adherence and its metastable on/off switching caused by phase fluctuation, which permits escape from locations where bactericidal host immune responses are strongest.
Heat shock protein 60 (Hsp60)
Physical stresses such as temperature, acidity and alkalinity can all triggered the synthesis of heat shock proteins, a family of highly preserved stress proteins that are widely present in both prokaryotes and eukaryotes. GroES-like HspA (Hsp10) and GroEL-like HspB (Hsp60) are the two Hsps, which are mainly produced by H. pylori. The increased production of Hsp60 at low pH, which interacts with sul-foglycolipid, a receptor-like sulfatide, indicates that acid stress may have an impact on the receptor selectivity of H. pylori.63 One of the expected immunogens of the bacterium’s that stimulates monocytes or gastric epithelial cells to release IL-6 and 8, tumor necrosis factor alpha (TNF-α), and GRO (Growth-Related Oncogene) has been identified as heat shock protein.64 Human monocytes release IL-8 when Hsp60 activates NF-κB through TLR2 and the mitogen-activated protein kinase pathway.65 Additionally, H. pylori-infected individuals consistently express anti-Hsp60 antibodies, and the titers are linked to the development of gastritis or stomach cancer.66 Further studies showed that through enhancing the production of IL-8, TNF-α, and monoclonal antibodies against H. pylori Hsp60 could influence bacterial pathogenesis.67 The purpose of the pathogen-specific antibodies is not to recognize or eradicate germs, but to carry out possible immunological functions. Although, more research work is required to clarify the function of anti-Hsp60 antibodies in H. pylori-related gastrointestinal disorders.
Outer membrane protein (OMP)
Nearly 65 OMPs, which fall within five gene families, are expressed by H. pylori. Out of 65 genes, genes in family 1 are called hor and hop. These are those genes which are coding for the adhesion proteins, like AlpA/B, SabA/B, BabA/B/C, etc. Besides these, two processes that regulate the synthesis of OMP are gene recombination and slipped strand mispairing, which take place in the dinucleotide repeat area near the 52-end of the particular gene.68
On the other hand, the five genes that make up the OMP family encode efflux pump proteins, which are essential for antibiotic resistance, thus finally helps in the infection process.69 There are several OMPs that have been found to contribute in the pathogenicity of H. pylori. They are playing crucial role to cause pathogenicity in the host’s epithelial cells.34
Toxin released and harm to the host tissue
H. pylori must be adhered to the epithelial cell and released some toxins after entering the stomach mucosa in order to initiate infection and also to damage the host cell. In this aspect, two most studied toxins released by H. pylori are cytotoxin-associated gene A (CagA) and vacuolating cytotoxin A (VacA). and cytotoxin-associated gene A (CagA).
Cytotoxin-associated gene A (CagA)
About 60% CagA-positive H. pylori infections occurs in European and North American countries, and 90% in Asian countries. Few studies showed that CagA-positive strains are directly belong to gastric ulcers and cancer and acute gastritis.70 Therefore, the pathogenicity of H. pylori strains can be quantified by their capacity to secrete CagA. By repeating the sequence of Glu-Pro-Ile-Tyr-Ala (EPIYA) motifs at the N-terminus of CagA, genes linked to cytotoxins enable the further classification of proteins into European and, North American type and East Asian-type CagA. The findings concluded that East Asian-type CagA generates greater morphological alterations and is more probable to be connected to stomach cancer.71
H. pylori’s chromosome contains the cag pathogenicity island i.e. cagPAI, which is 35-40 kb in size and associated with about 30 genes. Among these, cagPAI transports at least six genes that are homologous to type IV secretion systems (T4SS). The bacterial protein CagA is transferred into the host gastric cell’s cytoplasm when it comes into contact with epithelial cells. Additionally, a number of T4SS proteins, including CagL and CagY, use b1-integrin as a receptor to transport CagA into the host cell.72 The translocated CagA protein localizes to the inner surface of the plasma membrane through interactions with phosphatidylserine, where it is then tyrosine phosphorylated by a protein tyrosine kinase from the Src family. When CagA enters the cytoplasm through the T4SS, it can change host cell signaling in ways that are both phosphorylation-dependent and phosphorylation-independent. This binding of the phosphorylated CagA to the phosphatase SHP-2 affects the cell’s adhesion, migration, and spread.73 Additionally, CagA could affect the cell of the host in a number of ways, including the development of gastric epithelial cell pedestals, modifications to the cytoskeleton, effects on cell proliferation, and inducing the secretion of IL-8 from the epithelium of the gastric cells.74 Effects of the cytotoxin-associated gene A are phosphorylation independent, although many of these effects are still unknown. Recent research studies indicated that nonphosphorylated CagA’s C-terminus has a conserved motif that interacts with the host hepatocyte growth factor receptor to promote inflammation and cellular development by triggering the Akt signalling pathway, which in turn triggers NF-κB and β-catenin, according to recent studies.75
Vacuolating cytotoxin A (VacA)
Besides the Cag A cytotoxin released by H. pylori, VacA is the another toxin which is also released. Vacuolation is the process of forming vacuoles on a host cell. This process is used by pathogenic bacteria to prolong bacterial infections. Vacuolation caused by the actions of the VacA protein can have a number of harmful outcomes on its host cell, including cellular cytotoxicity and apoptosis.76 Additionally, it may cause MAP kinase activation, endocytic trafficking disruption, mitochondrial disruption and outflow of various ions, such as Cl, HCO3 and urea.77 The development of vacuoles on the cytoplasmic membrane makes the host’s stomach epithelial cell susceptible to urease action.
VacA is generated as 140 kDa precursor and then goes through a proteolytic process to create an 88 kDa toxin. It attaches itself to homologous receptors on epithelial cells, such as receptor-like protein tyrosine phosphatase alpha and beta (RPTP-α, RPTP-β), dense lipoprotein receptor-related protein-1 (LRP-1), and sphingomyelin. Additionally, it interacts with T cell CD18 receptors. After entering the extracellular space, the VacA toxin fragments (amino-terminal 33 kDa (p33) and carboxy-terminal 55 kDa (p55)) are internalized into endosomal compartments through a type V (autotransporter) secretion pathway when an amino-terminal signal peptide and a carboxy-terminal domain are present.78 VacA’s many polymorphic variants are linked to a number of clinical results.79
VacA, which the cell absorbs after being released by H. pylori. Endosomal compartments are associated with the absorbed VacA. Additionally, they have been linked to mitochondria, the endoplasmic reticulum, and the Golgi body.78 VacA can disrupt the normal function of the mitochondria by entering through the intracellular transporter system. Additionally, VacA can alter endocytic functions or intracellular trafficking, as well as prevent procathepsin D from maturing, disrupt the position of the transferrin receptor, and prevent mitochondrial fragmentation during antigen presentation. VacA may also alter intracellular trafficking or endocytic processes.78 Additionally, VacA proteins can disrupt the immune response by reducing or totally inhibiting T-lymphocyte activation in the lamina propria. Along with preventing the orderly breakdown and recycling of cellular components and the elimination of intracellular infections, it also prevents the immune system from eliminating damaged or malfunctioning cells. T cells, B cells, eosinophils, macrophages, dendritic cells, and neutrophils are among the immune cells that are affected.80 The process of apoptosis causes the host cells to destroy themselves, which regulates cell death. Cells that undergo this process often shrink and break into little pieces, which enhances their phagocytosis by neighbouring immune cells. Apoptosis rarely happens in healthy circumstances. Fortunately, an intricate system maintains a balance between cell development and death. When H. pylori are present, apoptosis is increased, especially in the host cells’ stomach gland. The vacA gene is present in all H. pylori strains, however the level of vacuolation activity of the cytotoxins produced differs.81 This variation is caused by internal duplications, deletions, nonsense mutations, or 1 bp insertions within the vacA gene.82 It has also been discovered that variations in the amino acid sequences,83 transcription, and efficiency of this gene secretion affect the amounts of vacuolating activity.84 The vacA gene has three sections: middle (m1 and m2), signal (s1 and s2), and intermediate (i1 and i2).85 The various vacA variants have pathogenic characteristics linked to each other.86
The toxins’ vacuolating action and cell surface binding comes from the middle portion.87 The signal region (sla, s1b, and s2) and middle region (m1 and m2) of the genome differ in sequence, leading to variation in mutational strains. The vacA s2/m2-carrying strains are completely non-cytotoxic, but the s1/m2-carrying strains are intermediately cytotoxic and the s1a/m1-carrying strains are highly cytotoxic. Additionally, vacA s1b/m1 have reduced activity.34 Based on WGS, the 41 H. pylori isolates were characterized in the research by Imkamp et al., to find the virulent genes cagA, vacA, iceA, and dupA, of which 19 (46.3%) possessed the vacA gene.34 A vacA s1 allele was found to be present in 23 individuals (or 56%). The vacA s1 allele was present in 15 out of 41 (36.6%) individuals (in 7 individuals; vacA sla/m1 and in 8 individuals; vacA sla/m2), and the vacA s1b allele in 8/41 (in 5 individuals; s1b/m1 and in 3 individuals; s1b/m2) whereas 18 isolates (43.9%) harboured the vacA s2/m2 allele. Additionally, a link was demonstrated between the development of gastritis and the presence of vacA s1 genes. Additionally, a strong association between vacA s1 and peptic ulcer disease was also discovered by Idowu et al.33 (Table).
Table
Some essential biological factors for pathogenicity of H. pylori in human
No. |
Biological factor |
Function |
Ref. |
|---|---|---|---|
1. |
Arginase |
Prevents the death of microbes. Stops the growth of T cells. Inhibit immune reactions. Increase apoptosis. Support H. pylori to survive in the acidic environment. |
88,89 |
2. |
Lipopolysaccharide |
Activate several signaling pathways. Cause a number of inflammatory reactions. Activate immunological reactions. Interferes with mucus secretion. Protect the body against harmful substances. |
90-92 |
3. |
Superoxide dismutase (SOD) |
Maintain reactive oxygen species (ROS) to enter the cell. To speed up colonization. Inhibits inflammatory cytokines production by encouraging neutrophils. |
93-96 |
4. |
Cholesteryl-α-glucosyl-transferase (aCgT) |
Protect Hostfrom immune attack. Increase the synthesis of IL-8. Increase bacterial proliferation and antibiotic resistance. |
97,98 |
5. |
ϒ-glutamyl-transferase |
Enhances necrosis and apoptosis. Promote the inflammatory proteins’ production cause ROS to be released. Promote the degradation of DNA. |
99,100 |
6. |
Phospholipase |
Increase in signaling channels like ERK1/2. Cause ongoing inflammation. Increase bacterial survival and colonization. Involved in lipid degradation and mucus layer harm. |
101,102 |
7. |
Duodenal ulcer‑promoting gene (dupA) |
The dupA gene produces a homolog, i.e. VirB4 ATPase. Particularly associated to gastric ulcers. DupA secrete pro-inflammatory cytokines. DupA has been recognized a biomarker for the gastric ulcers. |
103-105 |
8. |
High-temperature requirement A (HtrA) |
This protein can withstand high temperatures and pH. Function associated with the epithelial barrier is disrupted by HtrA. They target host cell factors. |
94 |
9. |
Catalase |
Catalase (KatA) is found in both plants and animal cells. It converts water (H2O) from hydrogen peroxide (H2O2). Participate in pathogenic processes, including inflammation, the inhibition of apoptosis, and the development of cancers caused by mutagenesis. |
106 |
Globally, H. pylori causes a huge number of cancers and ulcers in human beings and creates a high mortality rate. This pathogen creates effective colonization in the host gastric region with its adhesion molecules. It also produces some virulence protein/toxins responsible for various harmful human diseases. However, in the current scenario rising rates of infection, as well as the gastroduodenal pathological effects. For these reasons, the eradication of H. pylori may soon become a global concern. Furthermore, a number of methods for detecting and treating H. pylori infections are being developed in the present decade. The elevated incidence of H. pylori infection due to environmental contaminants present in the edible things and its effects on additional diseases make its eradication essential. For a complete understanding of the virulence variables and how they relate to the gastrointestinal illness, more research is required. Therefore, this review article could reveal important facets of diagnostic methods and mechanism of virulence in host. The crucial investigation on the virulence genes, mechanism and pathogenicity of H. pylori could be extremely valuable to pharmaceutical companies for the treatment of human chronic disorders associated with H. pylori.
ACKNOWLEDGMENTS
The authors would like to thank the Department of Biotechnology, Uttaranchal University, for giving valuable time to review the article and the Department of Medicine (Gastroenterology), Graphic Era Institute of Medical Science, Dhoolkot, Dehradun, Uttarakhand, India, for valuable suggestions.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
AUTHORS’ CONTRIBUTION
R performed literature review. MS, SDM, RSS and NS supervised the study. SR prepared tables and figures. R wrote the manuscript. MS, SDM and NS reviewed the manuscript. RSS edited the manuscript. All authors read and approved the final manuscript for publication.
FUNDING
None.
DATA AVAILABILITY
All datasets generated or analyzed during this study are included in the manuscript.
ETHICS STATEMENT
Not applicable.
- Donia MS, Fischbach MA. HUMAN MICROBIOTA. Small molecules from the human microbiota. Sci. 2015;349(6246):1254766.
Crossref - Duan Y, Xu Y, Dou Y, Xu D. Helicobacter pylori and gastric cancer: mechanisms and new perspectives. J Hematol Oncol. 2025;18(1):10.
Crossref - Kusters JG, van Vliet AHM, Kuipers EJ. Pathogenesis of Helicobacter pylori infection. Clin Microbiol Rev. 2006;19(3):449-490.
Crossref - Waskito LA, Yamaoka Y. The Story of Helicobacter pylori: Depicting Human Migrations from the Phylogeography. In: Kamiya, S., Backert, S. (eds) Helicobacter pylori in Human Diseases. Advances in Experimental Medicine and Biology (Vol. 11), vol 1149. Springer, Cham. 2019;1149:1-16.
Crossref - Malfertheiner P, Camargo MC, El-Omar E, et al. Helicobacter pylori infection. Nat Rev Dis Primers. 2023;9(1):19.
Crossref - Scholz KJ, Hohne A, Wittmer A, et al. Co-culture of Helicobacter pylori with oral microorganisms in human saliva. Clin Oral Invest. 2025;29(1):79.
Crossref - Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74-108.
Crossref - Queiroz DMM, Luzza F. Epidemiology of Helicobacter pylori infection. Helicobacter. 2006;11(Supl 1):1-5.
Crossref - Reshetnyak VI, Burmistrov AI, Maev IV. Helicobacter pylori: Commensal, symbiont or pathogen? World J Gastroenterol. 2021;27(7):545-560.
Crossref - Axon ATR. Review article Is Helicobacter pylori transmitted by the gastro oral route? Aliment Pharmacol Ther. 1995;9(6):585-588.
Crossref - Thomas JE, Gibson GR, Darboe MK, Weaver LT, Dale A. Isolation of Helicobacter pylori from human faeces. Lancet. 1992;340(8829):1194-1195.
Crossref - Calvet X, Lazaro MJR, Lehours P, Megraud F. Diagnosis and epidemiology of Helicobacter pylori Infection. Helicobacter. 2013;18(Suppl 1):5-11.
Crossref - Ofori EG, Adinortey CA, Bockarie AS, Kyei F, Tagoe EA, Adinortey MB. Helicobacter pylori infection, virulence genes’ distribution and accompanying clinical outcomes: The West Africa Situation. BioMed Res Int. 2019;2019(1):7312908.
Crossref - Reshetnyak VI, Reshetnyak TM. Significance of dormant forms of Helicobacter pylori in ulcerogenesis. World J Gastroenterol. 2017;23(27):4867-4878.
Crossref - Kao CY, Sheu BS, Wu JJ. Helicobacter pylori infection: An overview of bacterial virulence factors and pathogenesis. Biomed J. 2016;39(1):14-23.
Crossref - Sigal M, Rothenberg ME, Logan CY, et al. Helicobacter pylori activate and expands Lgr5+ stem cells through direct colonization of the gastric glands. Gastroenterol. 2015;148(7):1392-404.e21.
Crossref - Baj J, Forma A, Sitarz M, et al. Helicobacter pylori virulence factorsmechanisms of bacterial pathogenicity in the gastric microenvironment. Cell. 2020;10(1):27.
Crossref - Machlowska J, Baj J, Sitarz M, Maciejewski R, Sitarz R. Gastric cancer: epidemiology, risk factors, classification, genomic characteristics and treatment strategies. Int J Mol Sci. 2020;21(11):4012.
Crossref - Baj J, Korona-Glowniak I, Forma A, Maani A, Sitarz E, Rahnama-Hezavah M, Radzikowska E, Portincasa P. Mechanisms of the epithelial-mesenchymal transition and tumor microenvironment in Helicobacter pylori-induced gastric cancer. Cell. 2020;9(4):1055.
Crossref - Krzyek P. Helicobacter pylori Efflux Pumps: A Double-Edged Sword in Antibiotic Resistance and Biofilm Formation. Int J Mol Sci. 2024;25(22):12222.
Crossref - Sabbagh P, Javanian M, Koppolu V, Vasigala VR, Ebrahimpour S. Helicobacter pylori infection in children: an overview of diagnostic methods. Eur J Clin Microbiol Infect Dis. 2019;38(6):1035-1045.
Crossref - Munoz AB, Stepanian J, Solano-Gutierrez JS, Vale FF, Trespalacios-Rangel AA. Helicobacter pylori Infection in Colombia: Phylogeny, Resistome, and Virulome. Apmis. 2025;133(2):e70003.
Crossref - Marshall B, Adams PC. Helicobacter pylori: A Nobel pursuit? Can J Gastroenterol. 2008;22(11):895-896.
Crossref - Warren JR, Marshall B. Unidentified curved bacilli on gastric epithelium in active chronic gastritis. Lancet. 1983;1(8336):1273-1275.
Crossref - Tomb JF, White O, Kerlavage AR, et al. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nat. 1997;388(6642):539-547.
Crossref - Van der Merwe S. World gastroenterology organization global guideline: Helicobacter pylori in developing countries. J Clin Gastroenterol. 2011;45(5):383-388.
Crossref - Huang Y, Wang QL, Cheng DD, Xu WT, Lu NH. Adhesion and invasion of gastric mucosa epithelial cells by Helicobacter pylori. Front Cell Infect Microbiol. 2016;6:159.
Crossref - Yonezawa H, Osaki T, Kamiya S. Biofilm formation by Helicobacter pylori and its involvement for antibiotic resistance. Biomed Res Int. 2015;2015(1):914791.
Crossref - Hathroubi S, Hu S, Ottemann KM. Genetic requirements and transcriptomics of Helicobacter pylori biofilm formation on abiotic and biotic surfaces. Npj Biofilms Microbio. 2020;6(1):56.
Crossref - Hathroubi S, Servetas SL, Windham I, Merrell DS, Ottemann KM. Helicobacter pylori biofilm formation and its potential role in pathogenesis. Microbiol Mol Biol. 2018;82(2):e00001-18.
Crossref - Fauzia KA, Miftahussurur M, Syam AF, et al. Biofilm formation and antibiotic resistance phenotype of Helicobacter pylori clinical isolates. Toxins. 2020;12(8):473.
Crossref - Saxena A, Mukhopadhyay AK, Nandi SP. Helicobacter pylori: Perturbation and restoration of gut microbiome. J Biosci. 2020;45(1):110.
Crossref - Idowu A, Mzukwa A, Harrison U, P, et al. Detection of Helicobacter pylori and its virulence genes (cagA, dupA, and vacA) among patients with gastroduodenal diseases in Chris Hani Baragwanath Academic Hospital, South Africa. BMC Gastroenterol. 2019;19(1):73.
Crossref - Imkamp F, Lauener FN, Pohl D, et al. Rapid characterization of virulence determinants in Helicobacter pylori isolated from non-atrophic gastritis patients by next-generation sequencing. J Clin Med. 2019;8(7):1030.
Crossref - Davis R, Bryson HM. Levofloxacin: a review of its antibacterial activity, pharmacokinetics and therapeutic efficacy. Drugs. 1994;47(4):677-700.
Crossref - Dore MP, Osato MS, Realdi G, Mura I, Graham DY, Sepulveda AR. Amoxycillin tolerance in Helicobacter pylori. JAC. 1999;43(1):47-54.
Crossref - Xu C, Soyfoo DM, Wu Y, Xu S. Virulence of Helicobacter pylori outer membrane proteins: an updated review. Eur JClinMicrobiol Infect Dis. 2020;39(10):1821-1830.
Crossref - Ha NC, Oh ST, Sung JY, Cha KA, Lee MH, Oh BH. Supramolecular assembly and acid resistance of Helicobacter pylori urease. Nat Struct Biol. 2001;8(6):505-509.
Crossref - De Brito BB, da Silva FA, Soares AS, et al. Pathogenesis and clinical management of Helicobacter pylori gastric infection. World J Gastroenterol. 2019;25(37):5578-5589.
Crossref - Becker KW, Skaar EP. Metal limitation and toxicity at the interface between host and pathogen. FEMS Microbiol Rev.2014;38(6):1235-1249
Crossref - Ernst FD, Stoof J, Horrevoets WM, Kuipers EJ, Kusters JG, van Vliet AH. NikR mediates nickel-responsive transcriptional repression of the Helicobacter pylori outer membrane proteins FecA3 (HP1400) and FrpB4 (HP1512). Infect Immun. 2006;74(12):6821-6828.
Crossref - Fulkerson Jr JF, Mobley HL. Membrane topology of the NixA nickel transporter of Helicobacter pylori: two nickel transport-specific motifs within transmembrane helices II and III. J Bacteriol. 2000;182(6):1722-1730.
Crossref - Ansari S, Yamaoka Y. Helicobacter pylori virulence factor cytotoxin-associated gene A (CagA)-mediated gastric pathogenicity. Int J Mol Sci. 2020;21(19):7430.
Crossref - Olson JW, Maier RJ. Molecular hydrogen as an energy source for Helicobacter pylori. Sci. 2002;298(5599):1788-1790.
Crossref - Can F, Karahan C, Dolapci I, Demirbilek M, Tekeli A, Arslan H. Urease activity and urea gene sequencing of coccoid forms of H. pylori induced by different factors. Curr Microbiol. 2008;56(2):150-155.
Crossref - Elhariri M, Hamza D, Elhelw R, Hamza E. Occurrence of cagA+ vacA s1a m1 i1 Helicobacter pylori in farm animals in Egypt and ability to survive in experimentally contaminated UHT milk. Sci Rep. 2018;8:14260.
Crossref - Evans Jr DJ, Evans DG, Takemura T, et al. Characterization of a Helicobacter pylori neutrophil-activating protein. Infect Immun. 1995;63(6):2213-2220.
Crossref - Polenghi A, Bossi F, Fischetti F, et al. The neutrophil-activating protein of Helicobacter pylori crosses endothelia to promote neutrophil adhesion in vivo. J Immunol. 2007;178(3):1312-1320.
Crossref - Petersson C, Forsberg M, Aspholm M, et al. Helicobacter pylori SabA adhesin evokes a strong inflammatory response in human neutrophils which is down-regulated by the neutrophil-activating protein. Med Microbiol Immunol. 2006;195(4):195-206.
Crossref - Wang G, Hong Y, Olczak A, Maier SE, Maier RJ. Dual roles of Helicobacter pylori NapA in inducing and combating oxidative stress. Infect Immun. 2006;74(12):6839-6846.
Crossref - Amedei A, Cappon A, Codolo G, et al. The neutrophil-activating protein of Helicobacter pylori promotes Th1 immune responses. J Clin Invest. 2006;116(4):1092-1101.
Crossref - Ilver D, Arnqvist A, Ogren J, et al. Helicobacter pylori adhesin binding fucosylated histo-blood group antigens revealed by retagging. Sci. 1998;279(5379):373-377.
Crossref - Aspholm-Hurtig M, Dailide G, Lahmann M, et al. Functional adaptation of BabA, the H. pylori ABO blood group antigen binding adhesin. Science. 2004;305(5683):519-522.
Crossref - Chen MY, He CY, Meng X, Yuan Y. Association of Helicobacter pylori babA2 with peptic ulcer disease and gastric cancer. World J Gastroenterol. 2013;19(26):4242-4251.
Crossref - Sheh A, Chaturvedi R, Merrell DS, Correa P, Wilson KT, Fox JG. Phylogeographic origin of Helicobacter pylori determines host-adaptive responses upon coculture with gastric epithelial cells. Infect Immun. 2013;81(7):2468-2477.
Crossref - Backstrom A, Lundberg C, Kersulyte D, Berg DE, Boren T, Arnqvist A. Metastability of Helicobacter pylori bab adhesin genes and dynamics in Lewis b antigen binding. Proc Natl Acad Sci U S A. 2004;101(48):16923-16928.
Crossref - Nell S, Kennemann L, Schwarz S, Josenhans C, Suerbaum S. Dynamics of Lewis b binding and sequence variation of the babA adhesin gene during chronic Helicobacter pylori infection in humans. MBio. 2014;5(6):e02281-14.
Crossref - Sheu BS, Odenbreit S, Hung KH, et al. Interaction between host gastric Sialyl-Lewis X and H. pylori SabA enhances H. pylori density in patients lacking gastric Lewis B antigen. Am J Gastroenterol. 2006;101(1):36-44.
Crossref - Sheu BS, Sheu SM, Yang HB, Huang AH, Wu JJ. Host gastric Lewis’s expression determines the bacterial density of Helicobacter pylori in babA2 genopositive infection. Gut. 2003;52(7):927-932.
Crossref - Mahdavi J, Sonden B, Hurtig M, et al. Helicobacter pylori SabA adhesin in persistent infection and chronic inflammation. Science. 2002;297(5581):573-578.
Crossref - Alm RA, Ling LS, Moir DT, et al. Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature. 1999;397(6715):176-180.
Crossref - Kao CY, Sheu SM, Sheu BS, Wu JJ. Length of thymidine homopolymeric repeats modulates promoter activity of sabA in Helicobacter pylori. Helicobacter. 2012;17(3):203-209.
Crossref - Huesca M, Borgia S, Hoffman P, Lingwood CA. Acidic pH changes receptor binding specificity of Helicobacter pylori: a binary adhesion model in which surface heat shock (stress) proteins mediate sulfatide recognition in gastric colonization. Infect Immun. 1996;64(7):2643-2648.
Crossref - Lin CY, Huang YS, Li CH, et al. Characterizing the polymeric status of Helicobacter pylori heat shock protein 60. Biochem Biophys Res Commun. 2009;388(2):283-289.
Crossref - Zhao Y, Yokota K, Ayada K, et al. Helicobacter pylori heat-shock protein 60 induces interleukin-8 via a Toll-like receptor (TLR) 2 and mitogen-activated protein (MAP) kinase pathway in human monocytes. J Med Microbiol. 2007;56(Pt 2):154-164.
Crossref - Tanaka A, Kamada T, Yokota K, et al. Helicobacter pylori heat shock protein 60 antibodies are associated with gastric cancer. Pathol Res Pract. 2009;205(10):690-694.
Crossref - Liao KW, Lin CS, Chen WL, et al. Antibodies against Helicobacter pylori heat shock protein 60 aggravate HSP60-mediated pro-inflammatory responses. Cytokine. 2011;55(2):174-180.
Crossref - Marcus EA, Sachs G, Scott DR. Acid regulated gene expression of Helicobacter pylori: insight into acid protection and gastric colonization. Helicobacter. 2018;23(3):e12490.
Crossref - Bina JE, Alm RA, Uria-Nickelsen M, Thomas SR, Trust TJ, Hancock REW. Helicobacter pylori uptake and efflux: basis for intrinsic susceptibility to antibiotics in vitro. Antimicrob Agents Chemother. 2000;44(2):248-254.
Crossref - Sheu SM, Sheu BS, Yang HB, Li C, Chu TC, Wu JJ. Presence of iceA1 but not cagA, cagC, cagE, cagF, cagN, cagT, or orf13 genes of Helicobacter pylori is associated with more severe gastric inflammation in Taiwanese. J Formos Med Assoc. 2002;101(1):18-23.
- Argent RH, Kidd M, Owen RJ, Thomas RJ, Limb MC, Atherton JC. Determinants and consequences of different levels of CagA phosphorylation for clinical isolates of Helicobacter pylori. Gastroenterol. 2004;127(2):514-523.
Crossref - Tegtmeyer N, Lind J, Schmid B, Backert S. Helicobacter pylori CagL Y58/E59 mutation turns-off type IV secretion-dependent delivery of CagA into host cells. PLOS ONE. 2014;9(6):e97782.
Crossref - Yamazaki S, Yamakawa A, Ito Y, et al. The CagA protein of Helicobacter pylori is translocated into epithelial cells and binds to SHP-2 in human gastric mucosa. J Infect Dis. 2003;187(2):334-337.
Crossref - Boonyanugomol W, Chomvarin C, Baik SC, et al. Role of cagA-positive Helicobacter pylori on cell proliferation, apoptosis, and inflammation in biliary cells. Dig Dis Sci. 2011;56(6):1682-1692.
Crossref - Suzuki M, Mimuro H, Kiga K, et al. Helicobacter pylori CagA phosphorylation-independent function in epithelial proliferation and inflammation. Cell Host Microbe. 2009;5(1):23-34.
Crossref - Gebert B, Fischer W, Weiss E, Hoffmann R, Haas R. Helicobacter pylori vacuolating cytotoxin inhibits T lymphocyte activation. Science. 2003;301(5636):1099-102.
Crossref - Amieva M, Peek Jr RM. Pathobiology of Helicobacter pylori-induced gastric cancer. Gastroenterol. 2016;150(1):64-78.
Crossref - McClain MS, Beckett AC, Cover TL. Helicobacter pylori vacuolating toxin and gastric cancer. Toxins. 2017;9(10):316.
Crossref - Winter JA, Letley DP, Cook KW, et al. A role for the vacuolating cytotoxin, VacA, in colonization and Helicobacter pylori-induced metaplasia in the stomach. J Infect Dis. 2014;210(6):954-963.
Crossref - Raju D, Hussey S, Ang M, et al. Vacuolating cytotoxin and variants in Atg16L1 that disrupt autophagy promote Helicobacter pylori infection in humans. Gastroenterol. 2012;142(5):1160-1171.
Crossref - Wroblewski LE, Peek Jr RM, Wilson KT. Helicobacter pylori and gastric cancer: factors that modulate disease risk. Clin Microbiol Rev. 2010;23(4):713-739.
Crossref - Ito Y, Azuma T, Ito S, et al. Full-length sequence analysis of the vacA gene from cytotoxic and noncytotoxic Helicobacter pylori. J Infect Dis. 1998;178(5):1391-1398.
Crossref - Atherton JC, Cao P, Peek RM Jr, Tummuru MKR, Blaser MJ, Cover TL. Mosaicism in vacuolating cytotoxin alleles of Helicobacter pylori. Association of specific vacA types with cytotoxin production and peptic ulceration. J Biol Chem. 1995;270(30):17771-17777.
Crossref - Forsyth MH, Atherton JC, Blaser MJ, Cover TL. Heterogeneity in levels of vacuolating cytotoxin gene (vacA) transcription among Helicobacter pylori strains. Infect Immun. 1998;66(7):3088-3094.
Crossref - Rhead JL, Letley DP, Mohammadi M, et al. A new Helicobacter pylori vacuolating cytotoxin determinant, the intermediate region, is associated with gastric cancer. Gastroenterol. 2007;133(3):926-936.
Crossref - Sheikh AF, Yadyad MJ, Goodarzi H, et al. CagA and vacA allelic combination of Helicobacter pylori in gastroduodenal disorders. Microb Pathog. 2018;122:144-150.
Crossref - Wang HJ, Wang WC. Expression and binding analysis of GST-VacA fusions reveals that the C-terminal approximately 100-residue segment of exotoxin is crucial for binding in HeLa cells. Biochem Biophys Res Commun. 2000;278(2):449-454.
Crossref - Zhang J, Zhang X, Wu C, et al. Expression, purification and characterization of arginase from Helicobacter pylori in its apo form. PLOS ONE. 2011;6(10):e26205.
Crossref - Zhang X, Zhang J, Zhang R, et al. Structural, enzymatic and biochemical studies on Helicobacter pylori arginase. Int J Biochem Cell Biol. 2013;45(5):995-1002.
Crossref - Keenan JI, Davis KA, Beaugie CR, McGovern JJ, Moran AP. Alterations in Helicobacter pylori outer membrane and outer membrane vesicle-associated lipopolysaccharides under iron-limiting growth conditions. Innate Immun. 2008;14(5):279-290.
Crossref - Zimmermann S, Pfannkuch L, Al-Zeer MA, et al. ALPK1-and TIFA-dependent innate immune response triggered by the Helicobacter pylori type IV secretion system. Cell Rep. 2017;20(10):2384-2395.
Crossref - Yokota SI, Amano KI, Nishitani C, Ariki S, Kuroki Y, Fujii N. Implication of antigenic conversion of Helicobacter pylori lipopolysaccharides that involve interaction with surfactant protein D. Infect Immun. 2012;80(8):2956-2962.
Crossref - Morishita K, Takeuchi H, Morimoto N, et al. Superoxide dismutase activity of Helicobacter pylori per se from 158 clinical isolates and the characteristics. Microbiol Immunol. 2012;56(4):262-272.
Crossref - Svagelj D, Terzic V, Dovhanj J, Svagelj M, Cvrkovic M, Svagelj I. Superoxide dismutases in chronic gastritis. APMIS. 2016;124(4):252-256.
Crossref - Negovan A, Iancu M, Tripon F, Crauciuc A, Mocan S,
Banescu C. The CAT-262 C> T, MnSOD Ala16Val, GPX1 Pro198Leu polymorphisms related to oxidative stress and the presence of gastric lesions. J Gastrointestin Liver Dis. 2018;27(4):371-378.
Crossref - Stent A, Every AL, Chionh YT, Ng GZ, Sutton P. Superoxide dismutase from Helicobacter pylori suppresses the production of pro inflammatory cytokines during in vivo infection. Helicobacter. 2018;23(1):e12459.
Crossref - Morey P, Meyer TF. The sweeping role of cholesterol depletion in the persistence of Helicobacter pylori infections. Curr Top Microbiol Immunol. 2019;421:209-227.
Crossref - Morey P, Pfannkuch L, Pang E, et al. Helicobacter pylori deplete cholesterol in gastric glands to prevent interferon gamma Signaling and escape the inflammatory response. Gastroenterol. 2018;154(5):1391-1404.e9.
Crossref - Shibayama K, Wachino JI, Arakawa Y, Saidijam M, Rutherford NG, Henderson PJ. Metabolism of glutamine and glutathione via g-glutamyltranspeptidase and glutamate transport in Helicobacter pylori: possible significance in the pathophysiology of the organism. Mol Microbiol. 2007;64(2):396-406.
Crossref - Boonyanugomol W, Chomvarin C, Song JY, et al. Effects of Helicobacter pylori g-glutamyltranspeptidase on apoptosis and inflammation in human biliary cells. Dig Dis Sci. 2012;57(10):2615-2624.
Crossref - Sitaraman R, Israel DA, Romero-Gallo J, Peek Jr RM. Cell-associated hemolysis induced by Helicobacter pylori is mediated by phospholipases with mitogen-activated protein kinase-activating properties. J Clin Microbiol. 2012;50(3):1014-1018.
Crossref - Lusini P, Figura N, Valassina M, et al. Increased phospholipase activity in Helicobacter pylori strains isolated from patients with gastric carcinoma. Dig Liver Dis. 2005;37(4):232-239.
Crossref - Lu H, Hsu PI, Graham DY, Yamaoka Y. Duodenal ulcer promoting gene of Helicobacter pylori. Gastroenterol. 2005;128(4):833-848.
Crossref - Hussein NR, Argent RH, Marx CK, Patel SR, Robinson K, Atherton JC. Helicobacter pylori dupA is polymorphic, and its active form induces proinflammatory cytokine secretion by mononuclear cells. J Infect Dis. 2010;202(2):261-269.
Crossref - Pereira WN, Ferraz MA, Zabaglia LM, et al. Association among H. pylori virulence markers dupA, cagA and vacA in Brazilian patients. J Venom Anim Toxins incl Trop Dis. 2014;20:1-5.
Crossref - Switala J, Loewen PC. Diversity of properties among catalases. Arch Biochem Biophys. 2002;401(2):145-154.
Crossref
© The Author(s) 2025. Open Access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License which permits unrestricted use, sharing, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.