


ISSN: 0973-7510
E-ISSN: 2581-690X
The emergence of multidrug-resistant Mycobacterium tuberculosis (Mtb) strains has rendered many frontline antituberculosis agents ineffective, necessitating the urgent identification of novel therapeutic targets. One of the critical enzymes in Mtb lipid metabolism is enoyl-acyl carrier protein reductase (InhA), which catalyzes the NADH-dependent reduction of 2-trans-enoyl-ACP within the mycolic acid biosynthetic pathway. Inhibiting InhA with drugs blocks mycolic acid production, weakening the bacterial cell wall, disturbing metabolism, and ultimately lowering Mtb survival. Despite the existence of clinically approved InhA targeting agents, their therapeutic efficacy against drug-resistant Mtb strains is suboptimal, underscoring the necessity of identifying alternative inhibitors. Natural products, particularly phytochemicals derived from medicinal plants and herbs, represent a vast reservoir of structurally diverse bioactive molecules with potential antimicrobial properties. In this study, a structure-based virtual screening approach integrating molecular docking and molecular dynamics (MD) simulations was employed to identify potent phytochemical inhibitors of InhA. Chryso-obtusin (-8.92 kcal/mol), Episesamin (-8.74 kcal/mol), Apohyoscine (-7.84 kcal/mol), and Norhyosine (-7.74 kcal/mol)-exhibiting high-affinity interactions with the enzyme’s cofactor-binding domain. Subsequent MD simulations elucidated their stability and mechanistic similarity to isoniazid-mediated inhibition. These findings highlight the potential of natural phytochemicals as promising inhibitors of enoyl-ACP reductase, providing a foundation for the development of alternative therapeutic strategies against TB, particularly in the context of drug-resistant strains.
Mycobacterium Tuberculosis, Mycolic Acid, Himalayan Herbs, Molecular Docking, Molecular Dynamics
Tuberculosis (TB), an infectious disease caused by the obligate pathogen Mycobacterium tuberculosis (Mtb), remain a formidable global health challenge. In the post-COVID-19 era, TB continues to rank as the second most lethal infection-related pathology, accounting for significant morbidity and mortality worldwide.1 According to the WHO-TB (World Health Organization-TB) report, an estimated 10.6 million individuals were afflicted with TB in 2022, demonstrating an escalating trend from 10.3 million in 2021 and 10.0 million in 2020. Consequently, the disease led to 1.3 million fatalities in 2022, inclusive of 167,000 HIV co-infected individuals. A principal impediment in mitigating Mtb infections is the growing resistance to frontline anti-TB pharmacotherapeutics, culminating in the alarming prevalence of multidrug-resistant (MDR), extensively drug-resistant (XDR), and totally drug-resistant (TDR) strains.2
The cell wall of mycobacterial species is an essential part of their cell structure and plays a very important role in its survival and infection.3 It furnishes a robust protective barrier against antibacterial agents and facilitates the organism’s persistence in a quiescent, non-replicative state under adverse conditions.4,5 The mycobacterial cell wall comprises of three principal macromolecular constituents: mycolic acid, arabinogalactan, and peptidoglycan.3,6 Unlike other prokaryotes, mycobacteria take a unique approach to synthesizing fatty acids. They utilize both a eukaryote-like fatty acid synthase-1 (FAS-1) pathway and a prokaryotic-like fatty acid synthase-II (FAS-II) pathway for lipid synthesis.7 These pathways collectively contribute to the normal fatty acid synthesis as well as in the synthesis of the unique α-branched and β-hydroxylated mycolic acids. The highly hydrophobic nature of mycolic acid creates an impermeable envelope, rendering Mtb inherently recalcitrant to antimicrobial agents and significantly augmenting its virulence and persistence.8
The mycolic acid biosynthetic pathway has been proposed as a potential pharmacological target for the development of novel antimycobacterial agents, highlighting importance for Mtb pathogenesis and its exclusivity to bacterial physiology. Within the FAS-II system, key enzymatic players such as InhA, MabA (FabG1), KasA, and HadAB orchestrate the biosynthetic cascade, effectuating critical catalytic transformations in mycolic acid synthesis.9,10 The enzyme InhA is a short-chain reductase that catalyzes the NADH-specific reduction of 2-trans-enoyl-ACP in the final elongation step of the FAS-II system.10 Pharmacological inhibition of the enzyme leads to cell wall disintegration, fatty acid aggregation, and cell lysis, all of which contribute to the pathogen’s death.11
Despite the availability of InhA-targeting pharmacotherapeutics such as isoniazid (INH), ethionamide (ETA), and triclosan, these agents exhibit diminished efficacy against MDR and XDR Mtb strains. The majority of the drug-resistant clinical isolates exhibit resistant to INH, a key first-line anti-TB drug.12,13 Resistance is primarily attributed to mutations within the InhA promoter or coding region, which either upregulate enzyme expression or perturb the drug-binding interface, thereby abrogating INH-mediated inhibition.14,15 This necessitates the discovery of novel small-molecule inhibitors capable of circumventing resistance mechanisms and effectively targeting InhA functionality.
In this context, there is an emerging resurgence in the exploration of alternative therapeutic strategies as well as traditional medicinal practices. One such intervention is Hawan, an ancient Indian fumigation ritual involving the combustion of a polyherbal mixture.16,17 Historically, inhalation of the emanating bioactive volatiles has been claimed to exhibit antimicrobial properties, including anti-tubercular efficacy.18 Moreover, extracts from this mixture were used orally as medicine, as recommended in ancient Indian texts such as the Charak Samhita. Despite their ethnomedicinal prominence, they still remain underexplored within contemporary biomedical research due to a lack of experiential validation. However, recent studies have begun to investigate the detoxifying and antibacterial attributes of Hawan,19 with preliminary in vitro assessments demonstrating antitubercular activity against specific Mtb strains.18,20
In this study, we performed a structure-based virtual screening of 37 unreported phytochemicals derived from 27 Himalayan herbs and spices traditionally used in Hawan samagri. This study aimed to identify novel inhibitory scaffolds targeting the enoyl-acyl carrier protein reductase enzyme of Mtb, for their potential application in the treatment of drug-resistant TB.
Compound selection and target identification
Screening of compounds was done by searching the available literature and Dr. Duke’s Phytochemical and Ethnobotanical Databases.14 We selected 37 compounds from 27 Himalayan herbs and spices commonly used as hawan samagri in India.18 The names of these plants and selected compounds are given in Supplementary Table 1. All these compounds are of small in size and have a molecular weight between 300 and 600 Dalton. For molecular docking, the three-dimensional structures of these phytochemicals/compounds were downloaded from the PubChem database (https://pubchem.ncbi.nlm.nih.gov/) in SDF format. The downloaded SDF files were converted into PDB files using BIOVIA Discovery Studio Visualizer 2021 (BIOVIA, Dassault Systemes; https://discover.3ds.com/discovery-studio).
Drug-likeness analysis
The selected phytochemicals were screened for their drug properties using Lipinski’s rule of 521 (http://www.scfbio-iitd.res.in) (Table 1 and Supplementary Table 2). For the study of drug properties, compounds with standard parameters including hydrogen bond donors (<5), hydrogen bond acceptors (<10), high lipophilicity (LogP <5), molecular mass (<500 Daltons), and molar refraction (40-130) were selected.
Table (1):
Lipinski profiling of selected phytochemicals
No. |
Compounds |
Molecular Mass (g/mol) (≤500) |
Xlog P3 (
| H donor (≤5) |
H acceptor (≤10) |
Molar Refractivity (40-130) |
|---|---|---|---|---|---|---|
1. |
Chryso-obtusin |
358 |
2.6 |
1 |
7 |
85.5 |
2. |
Episesamin |
354 |
3.0 |
0 |
6 |
87.3 |
3. |
Norhyoscine |
289 |
1.4 |
2 |
5 |
73.6 |
4. |
Apohyoscine |
285 |
1.8 |
0 |
3 |
74.5 |
ADME/Toxicity analysis
The ADME/Tox profile is the main reason for the failure of many drug candidates in clinical trials. For this reason, we use the online server PreADMET (http://preadmet.bmdrc.org/, accessed on October 12, 2021) to screen pharmacokinetic attributes such as human intestinal absorption (HIA), Caco-2 in vitro cell permeability (PCaCO2), Maden-Darby Canine Kidney (PMDCK) cell permeability, skin permeability (PSkin), plasma Protein Binding (PPB), blood-brain barrier diffusion (CBrain/CBlood), and toxicological attributes such as mutagenicity and carcinogenicity. Those compounds matching the ADME/T parameters were further subjected to docking and simulation analysis22 (Table 2 and Supplementary Table 3).
Table (2):
ADMET profiling of the selected phytochemicals
No |
Compounds |
Mutagenicity (Ames Test) |
Carcinogenicity |
HIA % |
Pcaco-2 (nm/s) |
Pmdck (nm/s) |
Pski (nm/s) |
PPB % |
BBB % |
|---|---|---|---|---|---|---|---|---|---|
1. |
Chryso-obtusin |
Mutagen |
Non-carcinogenic |
96.19 |
22.34 |
175.63 |
-3.72 |
85.69 |
0.01 |
2. |
Episesamin |
Mutagen |
Non-carcinogenic |
97.95 |
57.02 |
20.60 |
-4.42 |
83.12 |
0.05 |
3. |
Norhyoscine |
Mutagen |
Non-carcinogenic |
92.06 |
20.49 |
1.41 |
-4.66 |
18.72 |
0.02 |
4. |
Apohyoscine |
Mutagen |
Non-carcinogenic |
98.02 |
53.50 |
49.45 |
-3.96 |
35.00 |
0.02 |
Preparation of proteins and ligands
The three-dimensional structure of InhA in combination with NAD, which serves as a cofactor, was downloaded from the RCSB database (PDB ID 1BVR) at 2.80 Å resolution for structure-based virtual screening.23 This structure initially includes multiple chains and NAD cofactor bound in it. For docking studies, only one chain was selected and heteroatoms, water molecules, and ligands (NAD cofactor) were removed from structures by using BIOVIA Discovery Studio Visualizer 2021 client (BIOVIA, Dassault Systemes; https://discover.3ds.com/discovery-studiovisualizer-download.
The three-dimensional structures of the selected ligands were retrieved in SDF format from the PubChem database (https://pubchem.ncbi.nlm.nih.gov/compound; accessed on 21 May 2022). These structures were then processed using BIOVIA Discovery Studio Visualizer to convert the SDF files into PDB format. Subsequently, the PDB files were imported into AutoDock 4.2.6, where they were converted into the PDBQT format for molecular docking studies.
Structure-based virtual screening
To prepare the target protein for structure-based virtual screening, a number of important steps were taken to ensure its suitability for virtual screening. The processed structures of the ligands and molecules were then subjected to molecular docking studies using AutoDock 4.2 software (AutoDock 4.2, Scripps Research, La Jolla, CA, USA, https://autodock.scripps.edu/downloaded, viewed on October 17, 2023).24 The Lamarckian genetic algorithm was used to estimate the binding energy and the inhibition constant of the interaction between the ligands and the targets (a maximum of 10 runs were performed for each compound to obtain optimal runs).25-27 For the docking analysis, we used a blind docking approach in which we selected the entire surface of a protein without specifying the target pocket. We created a grid of size 70 × 70 × 70 with coordinates 5.789, 25.971, and -4.844 to cover the entire surface of the InhA protein. For all upcoming interaction studies, the docked complex in the cluster with the lowest energy was evaluated. Both the inhibition constant (Ki) and binding energy are calculated and presented in Table 3 and Supplementary Table 3. Compounds were scored based on their binding energy, with the top 4 phytochemical compounds selected based on their ability to form the most favourable interactions with the target protein InhA.
Table (3):
Binding energy, inhibition constant, and interacting amino acid information of selected phytochemicals
No |
Compounds |
Plants |
Plant Family |
Plant Part |
Class of Compounds |
B.E. (kcal/mol) |
Ki (μM) |
Interacting amino acid |
Bond Length |
|---|---|---|---|---|---|---|---|---|---|
1. |
Chryso-obtusin |
Cassia tora |
Fabaceae |
Seed |
Polyketides
|
-8.92 |
0.28 |
VAL65, LEU63, ASP64, PHE41, ILE95, ILE122, GLY96, ILE16, PHE97, GLN66, SER94, GLY14 |
3.32 4.25 |
2. |
Episesamin |
Cinnamomum camphora |
Lauraceae |
Leaves |
Phenylpropanoids |
-8.74 |
0.35 |
ALA157, TYR158, PRO193, PHE149, ILE21, THR196, SER94, ILE194, MET199, LEU218, PRO156 |
5.63 4.19 |
3. |
Norhyoscine |
Datura metel |
Solanaceae |
Root |
Tropane alkaloid |
-7.84 |
1.78 |
TYR158, ASP148, MET147, PRO193, PHE149, MET199, ILE194, ILE21, GLY192, ALA191 |
4.95 4.54 5.23 |
4. |
Apohyoscine |
Datura metel |
Solanaceae |
Root |
Tropane alkaloid |
-7.74 |
2.13 |
PRO156, ALA157, MET155, TYR158, ILE215, LEU218, MET103, GLY192, ILE194, MET199, PHE149, PRO193 |
4.07 |
5. |
Isoniazid |
– |
– |
– |
Anti-TB drug |
-5.49 |
549.74 |
ALA198, THR196, PHE149, PRO193, TYR158, MET103, MET161, MET199 |
6.99 |
Molecular dynamics simulation
Molecular dynamics (MD) simulation of the top four docked protein-ligand complexes was performed to analyze the dynamic stability and intermolecular interactions at 50 ns using the Cresset academic package (Flare) V6.0 tool of Cresset Software.28 In the first step of the MD simulation, the protein-ligand complexes were opened in PDB file format in the working window of the flare tool and protein was prepared using the protein preparation tool.29 In the next step, the following criteria were selected for the energy minimization of the complex: Calculation method-GAFF2 with explicit water; custom parameter: A-chain, Simulation length- 50 ns, Solvation model: explicit; water model: explicit TIP3P water, Localho: 56245, and a total of 5000 frames and 2085 force-field parameters were saved. After running MDS, a DCD trajectory file is created, When we open the DCD file, we get all the parameters such as root mean square deviation (RMSD), 2D-dimensional root mean square deviation (2D RMSD), root mean square fluctuation (RMSF), radius of gyration (RG), properties (potential energy, temperature, box volume, density, speed), secondary structure, contacts, clustering, measurements.30 The docked complexes of all four compounds were individually subjected to MD simulations for durations of 50 to 200 ns, with an extended 200 ns simulation specifically conducted for the Chryso-obtusin-InhA complex, while the remaining ligand-protein systems were analyzed for 50 ns.
Screening of Himalayan herbs and spices to identify potential compounds
The primary objective of this study was to identify potent bioactive compound(s) from natural sources that could inhibit InhA, a key enzyme in Mtb, for the development of novel anti-TB drugs. Based on literature surveys and ethnobotanical documentation, we selected 27 plant species widely recognized for their historical and cultural significance in Hawan practices. These included Allium sativum, Albizia lebbeck, Punica granatum, Allium cepa, Ageratum conyzoides, Areca catechu, Butea monosperma, Commiphora mukul, Cinnamomum camphora, Cassia tora, Cannabis sativa, Capsicum annuum, Centella asiatica, Capsicum annuum, Datura metel, Elettaria cardamomum, Myristica fragrans, Punica granatum, Nigella sativa, Justicia adhatoda, Verbascum thapsus and Zingiber officinale. These plants are known to contain a diverse array of bioactive phytochemicals, several of which have been studied for their pharmacological properties. However, the specific roles of the phytochemicals isolated from these plants in targeting Mtb remain largely unexplored. To systematically screen for potential anti-TB compounds, we utilized Dr. Duke’s Phytochemical and Ethnobotanical Database, a comprehensive repository of plant-derived bioactive compounds.31 From this dataset, we identified 37 phytochemical compounds present in the selected plant species (Supplementary
Table 1). The subsequent analysis aimed to evaluate these compounds for their potential inhibitory activity against InhA, thus providing insights into their possible therapeutic applications in TB treatment.
Drug similarity and ADMET profiling
In drug discovery, phytochemical compounds proposed as therapeutic candidates must have both significant biological activity and negligible to minimal toxicity. As a result, a number of essential pharmacological characteristics, including drug-likeness and ADMET parameters (absorption, distribution, metabolism, excretion, and toxicity), have been established for the validation of each potential drug candidate.32 Pre-evaluation of these properties in the early stages of drug discovery is critical to prevent pharmacokinetic-related failures of pharmacologic agents in clinical trials.
The drug-like characteristic of selected phytochemicals was evaluated using Lipinski Rule of Five, a widely accepted criterion for assessing oral bioavailability in humans.21 According to this rule, a compound is deemed orally active if it satisfies the following parameters: a maximum of 10 rotatable bonds, no more than 10 hydrogen bond acceptors (HBA), fewer than 5 hydrogen bond donors (HBD), and a calculated partition coefficient Xlog P (which indicates acceptable lipophilicity), not exceeding 5. Phytochemicals that adhere to these physicochemical constraints exhibit enhanced permeability and bioavailability, thereby facilitating their interaction with the target receptor (Supplementary Table 2). We have selected 4 top compounds which follow Lipinski rule of five. Chryso-obtusin has a molecular mass of 358 g/mol, XlogP of 2.6, 1 hydrogen bond donor, 7 acceptors, and a molar refractivity of 85.5 all within the acceptable range. Episesamin, with a mass of 354 g/mol, XlogP of 3.0, no hydrogen bond donors, 6 acceptors, and molar refractivity of 87.3, also complies fully. Norhyoscine (289 g/mol) and Apohyoscine (285 g/mol) similarly meet all the criteria, with low molecular weights, low XlogP values, and acceptable numbers of hydrogen bond donors and acceptors, and molar refractivities within the required range. The specific drug-likeness attributes of the analyzed compounds are presented in Table 1.
Additionally, a comprehensive drug-likeness evaluation was conducted for all ligands within the examined compound library. The druggability assessment outcomes for these 37 compounds are systematically cataloged as supplementary data in Supplementary Table 3.
The ADME/Tox profiling of all selected phytochemicals was computationally predicted using the PreADMET online server (http://pread-met.bmdrc.org/). We have selected 4 top compounds which follow ADME/Tox profiling (Table 2). In terms of human intestinal absorption (HIA%), all compounds showed high absorption, with Apohyoscine at 98.02%, Episesamin at 97.95%, Chryso-obtusin at 96.19%, and Norhyoscine at 92.06%. Caco-2 permeability, which indicates intestinal absorption, ranged from 20.49 nm/s (Norhyoscine) to 57.02 nm/s (Episesamin), showing moderate permeability. For MDCK permeability, Chryso-obtusin showed the highest value at 175.63 nm/s, followed by Apohyoscine (49.45 nm/s), Episesamin (20.60 nm/s), and Norhyoscine (1.41 nm/s). The skin permeability (Pski) values were all negative, with Chryso-obtusin having the highest permeability at -3.72. Plasma protein binding (PPB%) varied widely, with Chryso-obtusin and Episesamin showing high binding rates at 85.69% and 83.12%, respectively, while Norhyoscine and Apohyoscine had lower PPB values of 18.72% and 35.00%. Blood-brain barrier (BBB%) penetration was minimal for all compounds, with values ranging from 0.01% to 0.05%, indicating limited central nervous system exposure.
Toxicological prediction results are presented in Table 2. Based on the pharmacokinetic profile and drug score, the top five compounds were selected, and molecular modeling studies were performed.
Molecular docking analysis
In the mycolic acid biosynthetic pathway, InhA catalyzes the terminal elongation step of the fatty acid synthase II (FAS-II) system, a process essential for the biosynthesis of mycolic acid.33 The substrate binding domain of InhA structurally delineated into three distinct sites: Site I, the catalytic domain, Site II, a hydrophobic pocket that accommodates fatty acid chains, and Site III, a solvent-exposed region.34 Additionally, InhA harbors a cofactor-binding pocket that specifically interacts with NAD. Structural analyses of this site reveal an abundance of charged residues that mediate NAD stabilization through a network of hydrogen bonding and p-p stacking interactions (Figure 1A and 1B).
An example of a potent InhA inhibitor is (INH), which binds to the substrate binding site in this pocket, forms a covalent adduct with NAD, and occupies both the NAD binding site and the substrate binding site, thus acting as a competitive inhibitor of both NADH and substrate binding to InhA35 (Figure 1C). This formation of the INH-NAD complex effectively disrupts enzymatic function, leading to bacteriostatic effects.
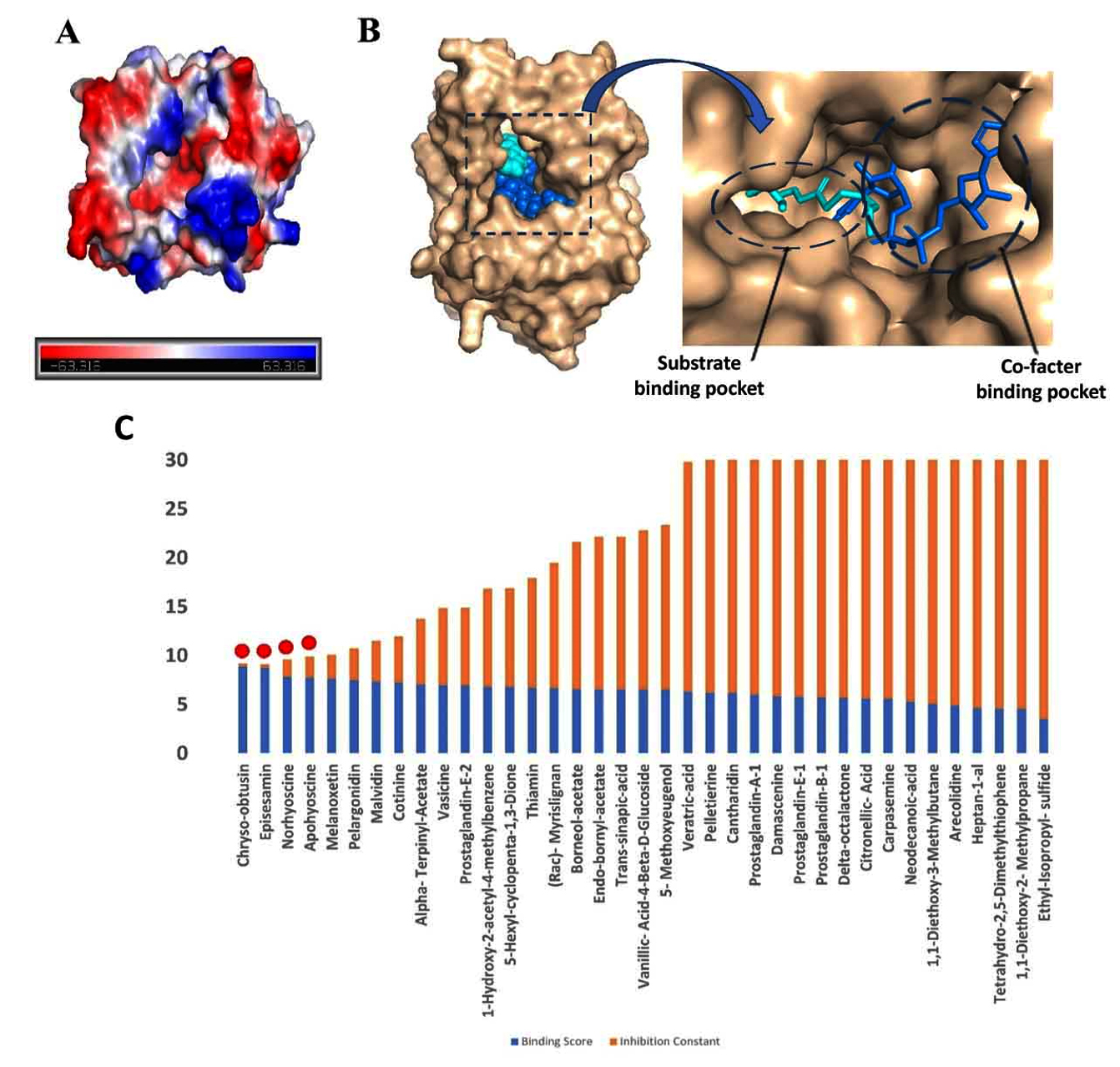
Figure 1. Molecular docking studies: (A) The surface electrostatic potential map of InhA showing cofactor binding site surrounded by charged residues. The blue color represents positive charge, whereas the red color represents negative charge residues. (B) Surface representations of the InhA structure (wheat) bound to NAD (blue) at the co-factor and INH at the substrate binding site (cyan). (C) A bar graph displaying molecular docking data for 37 herbal compounds, where blue represents binding energy and orange represents inhibition score. The four possible compounds chosen for molecular docking are outlined in red dots
Besides having reference information on the INH binding site in the InhA protein structure,23 we still selected the blind docking approach to comprehensively assess the specificity of ligand interactions with InhA. Molecular docking results revealed that the majority of the tested phytochemicals preferentially occupied the similar substrate-binding pocket as INH, a comparable inhibitory mechanism. The docking results are presented in terms of binding energy (BE) and inhibition constant (Ki) in Table 3 and Supplementary Table 4.
Based on binding affinity and toxicity assessments, four compounds-Chryso-obtusin, Episesamin, Norhyoscine, and Apohyoscine-emerged as lead candidates with significant inhibitory potential. A detailed interaction analysis of these top-ranked ligands is illustrated in Figure 2 (A-D) and Figure 3 (A-D).
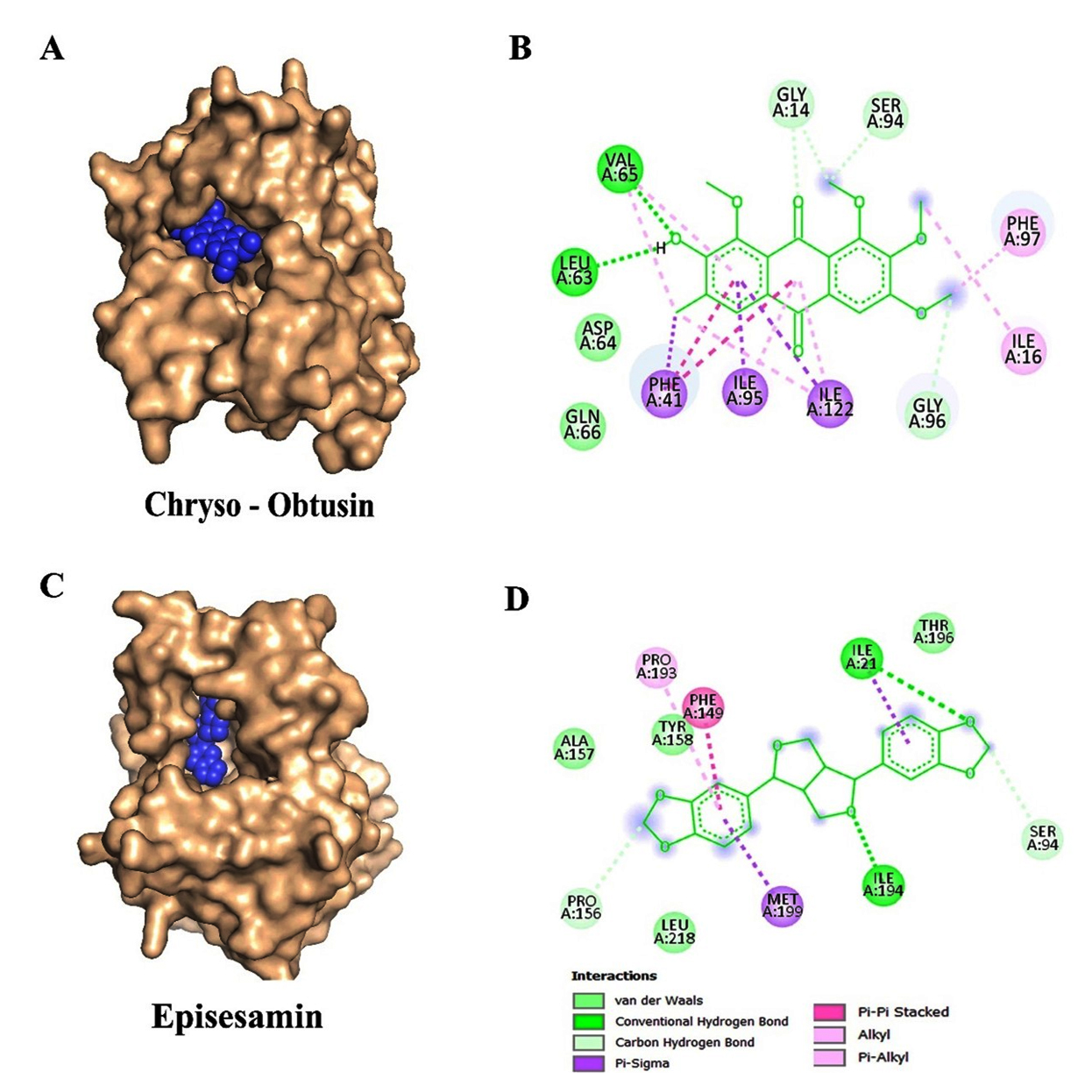
Figure 2. Molecular docking interactions of Chryso-obtusin and Episesamin with the target protein: (A) Surface representation showing the binding pocket of the protein with Chryso-obtusin (blue spheres) docked into the active site. (B) 2D interaction of Chryso-obtusin showing key residues involved in binding, including van der Waals interactions (lime green), hydrogen bonds (green), and various hydrophobic contacts such as Pi-Pi stacked, Pi-alkyl, and alkyl interactions (purple and pink). (C) Surface representation of the protein showing the binding of Episesamin (blue spheres) at the active site. (D) 2D interaction of Episesamin depicting van der Waals, hydrogen bonding, and hydrophobic interactions with active site residues
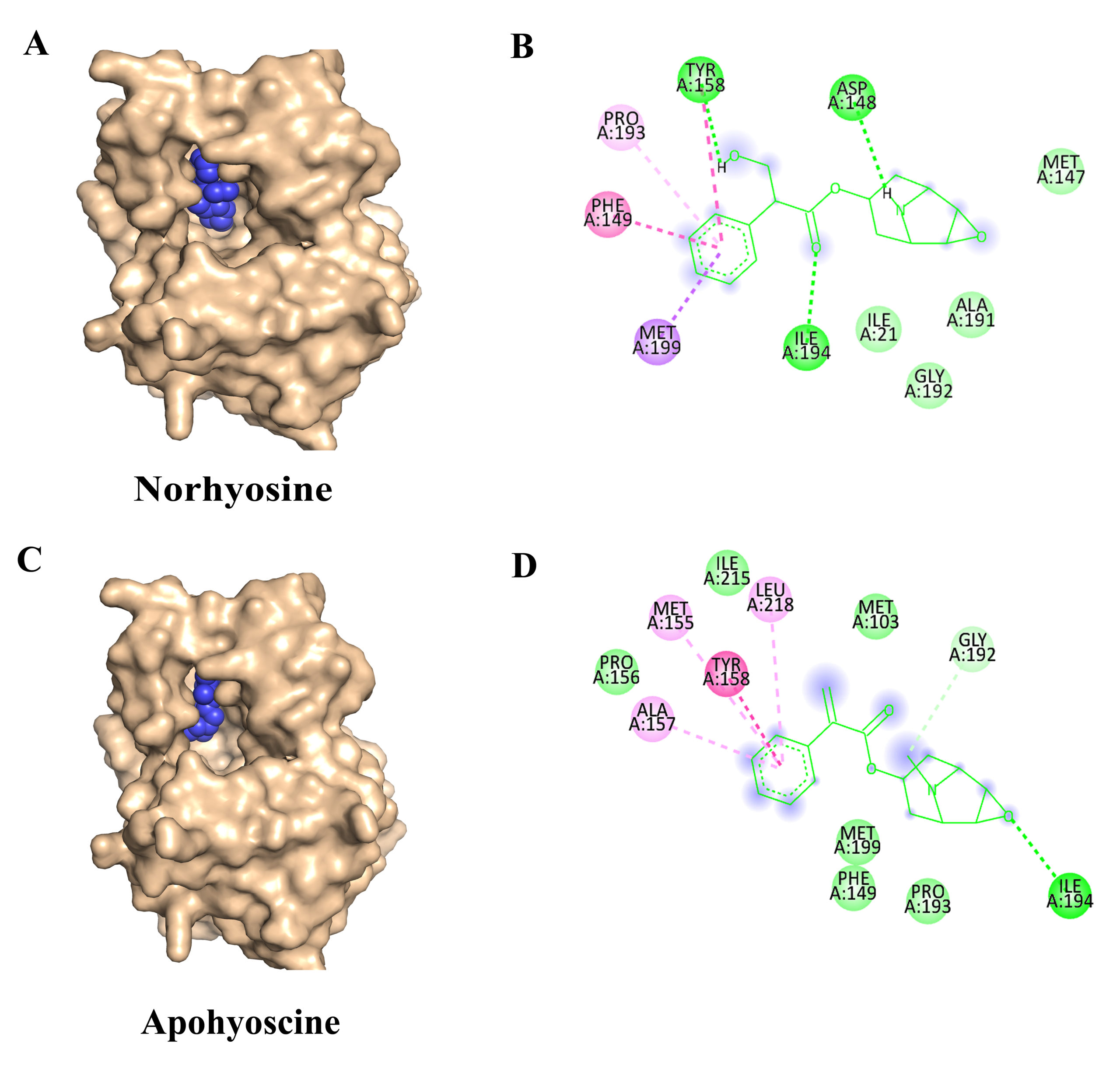
Figure 3. Molecular docking interactions of Norhyoscine and Apohyoscine with the target protein: (A) Surface representation of the protein (wheat) showing the binding of Norhyoscine (blue spheres) within the active site pocket. (B) 2D interaction of Norhyoscine highlighting its interactions with surrounding amino acid residues, including van der Waals contacts (lime green), hydrogen bonds (green), and hydrophobic interactions such as Pi-Pi stacked, Pi-alkyl, and alkyl (pink and purple). (C) Surface representation of the protein with Apohyoscine (blue spheres) bound at the active site. (D) 2D interaction of Apohyoscine showing various non-covalent interactions with active site residues, including van der Waals, hydrogen bonding, and hydrophobic contacts
Our molecular docking results highlight that Chryso-obtusin is the most potent InhA
inhibitor amongst the tested phytochemicals, exhibiting a binding free energy of -8.92 kcal/mol and an inhibition constant (Ki) of 289.31 nM. Chryso-obtusin, a mono-hydroxyanthraquinone derived from Cassia tora plant.36 Docking analysis shows that it forms hydrogen bonds with residues Val65 and Leu63, alongside a p-p stacking interaction with Phe97 of the InhA protein (Figure 2A and 2B).
The second-highest binding affinity was observed for Episesamin, a natural lignan isolated from Commiphora mukul. This compound demonstrated a binding free energy of -8.74 kcal/mol and a Ki of 395.09 nM. Structural analysis revealed that Episesamin forms hydrogen bonds with Ile21 and Ile194, while also engaging in p-p interactions with Phe149 (Figure 2C and 2D).37
The third-ranked inhibitor, Norhyoscine, displayed a binding free energy of -7.84 kcal/mol and an inhibition constant (Ki) of 1.78 µM.38 This tropane alkaloid, a derivative of norscopolamine found in Datura metel, interacts with InhA via hydrogen bonding with Tyr158, Asp148, and Ile194, while forming a p-p interaction with Phe149 (Figures 3A and 3B).
The fourth identified inhibitor, Apohyoscine, exhibited a binding free energy of -7.74 kcal/mol and an inhibition constant (Ki) of 2.13 µM.39 This tropane alkaloid, also derived from Datura metel, establishes hydrogen bonding interactions with Ile194, while engaging in a p-p stacking interaction with Tyr158 (Figures 3C and 3D).
Binding Site Specificity and Mechanistic Implications
In Figure 4A, the NAD-InhA complex highlights the cofactor-binding groove of InhA, which includes the adenine-binding pocket of NAD, critical for NAD cofactor association. Figure 4B shows that Chryso-obtusin demonstrates a unique binding orientation compared to other ligands studied, associating near this cofactor-binding region of InhA. It occupies a position that overlaps with the adenine-binding pocket of NAD, part of the larger NADH-binding groove. Chryso-obtusin forms multiple interaction with residues such as ILE95, PHE97, VAL65, ALA122, and GLN66, stabilizing its conformation within this cofactor-binding pocket. By sterically occupying the adenine moiety region of NAD, Chryso-obtusin likely hinders the natural cofactor from properly binding to InhA, thus inhibiting enzymatic activity through a non-competitive or allosteric mechanism.
In Figure 4C, the anti-tubercular drug INH binds at a similar location within the substrate-binding site. Upon activation by the KatG enzyme, INH forms a covalent adduct with NAD, leading to irreversible inhibition of InhA by blocking NADH access. This covalent inhibition mechanism forms the cornerstone of INH’s bactericidal action. In Figure 4C, the anti-tubercular drug INH binds at a similar location within the substrate-binding site. Upon activation by the KatG enzyme, INH forms a covalent adduct with NAD, leading to irreversible inhibition of InhA by blocking NADH access. This covalent inhibition mechanism forms the cornerstone of INH’s bactericidal action. Structural analyses of Episesamin (Figure 4D), Norhyoscine (Figure 4E), and Apohyoscine (Figure 4F) reveal that these compounds predominantly localize within the substrate-binding pocket of InhA, a deeply buried hydrophobic cleft highly conserved among mycobacterial strains.
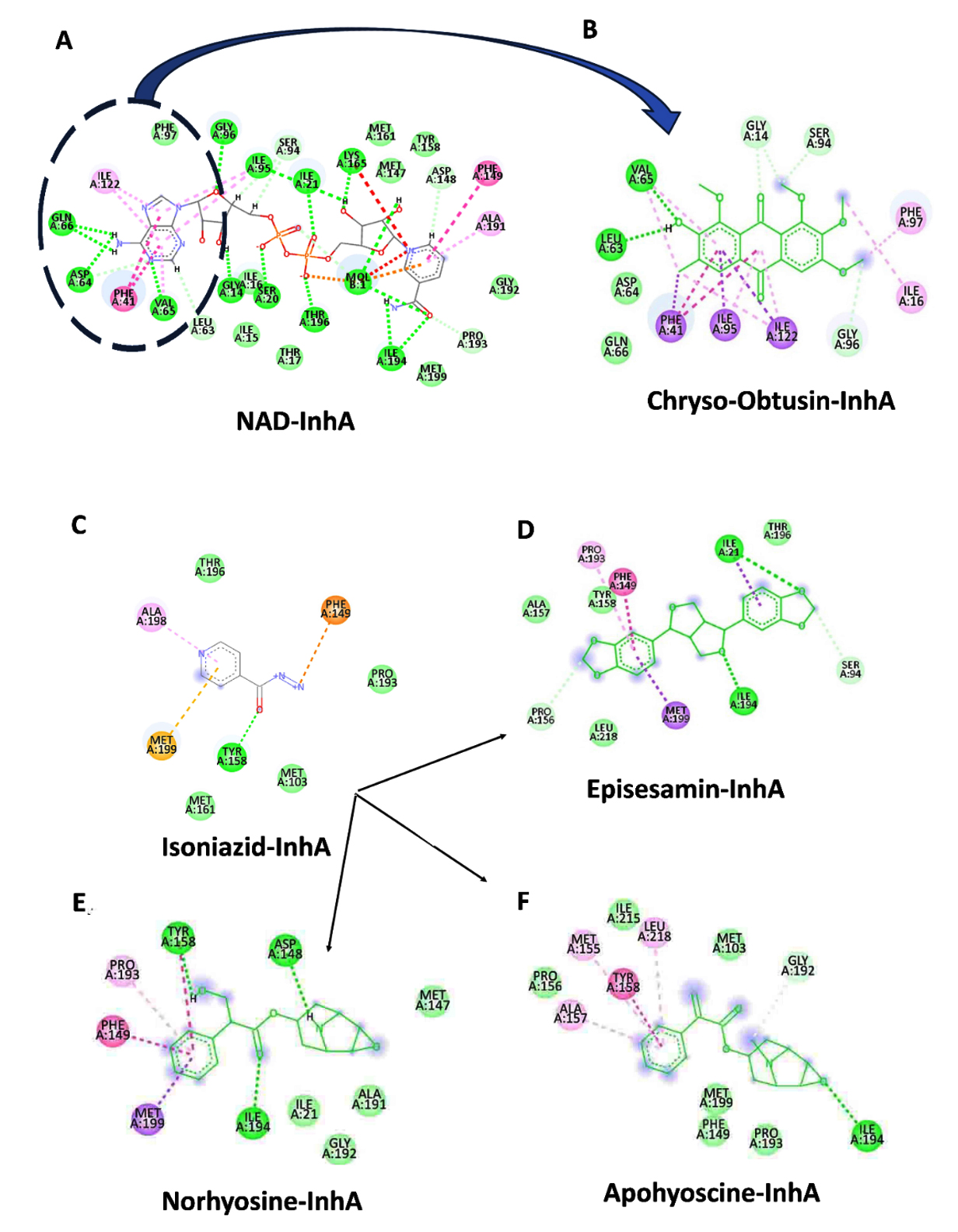
Figure 4. 2D interaction diagrams of NAD, isoniazid, and selected phytochemicals with InhA enzyme from Mtb: (A) Binding interaction of the NAD cofactor with InhA, highlighting the adenine-binding region (circled in blue), which is a critical site for enzymatic activity. (B) Chryso-obtusin binds in close proximity to the cofactor-binding region, occupying the adenine moiety site of NAD, suggesting a potential non-competitive or allosteric inhibitory mechanism that may block NAD association with InhA. (C) Isoniazid (INH), following activation, forms a covalent adduct with NAD and occupies the substrate-binding site of InhA, effectively obstructing NADH access. (D-F) The phytochemicals Episesamin (D), Norhyoscine (E), and Apohyoscine (F) predominantly localize within the substrate-binding pocket, interacting with conserved hydrophobic residues such as Tyr158, Phe149, Met199, Leu218, and Pro193. Their spatial alignment with the INH binding site suggests a similar competitive inhibition mechanism targeting NADH binding. These distinct binding modes illustrate the potential of Chryso-obtusin as a cofactor-binding inhibitor and Episesamin, Norhyoscine, and Apohyoscine as substrate-competitive inhibitors, supporting a dual-site therapeutic strategy for novel InhA-targeted TB treatment
This pocket is primarily formed by critical residues including Tyr158, Phe149, Met199, Try222, Leu218, Met161, and Pro193, many of which are also involved in the binding of NADH and the fatty acyl substrate. The binding profiles of Episesamin, Norhyoscine, and Apohyoscine reveal strong overlap with the binding mode of INH, both in terms of spatial orientation and residue interactions. This structural and energetic alignment suggests that these phytochemicals may function via a substrate-competitive inhibition mechanism, wherein they mimic the INH-NAD adduct by preventing NADH from engaging with the active site.
Taken together, this dual-targeting paradigm-with Chryso-obtusin acting near the cofactor-binding site and the other phytochemicals targeting the substrate-binding cleft-offers a compelling strategy for multi-site inhibition of InhA. The unique binding behavior of Chryso-obtusin, distinct from the classic substrate-competitive inhibition seen with INH and its analogs, raises the intriguing possibility of designing synergistic drug combinations. Such a regimen could pair Chryso-obtusin or its derivatives with INH or similar agents to simultaneously block cofactor and substrate binding, potentially overcoming drug resistance and enhancing therapeutic efficacy. This opens new avenues for developing next-generation InhA inhibitors with novel binding mechanisms tailored for improved activity and resistance management in TB treatment.
To assess whether these phytochemicals interfere with NAD binding to InhA, molecular docking was performed using the NAD-bound enzyme structure. INH, the reference anti-tubercular drug (Figure 5A), bound firmly within the conserved substrate-binding pocket, engaging catalytic residues such as Tyr158, Phe149, Met199, Leu218, and Thr196-residues also involved in stabilizing the INH-NAD adduct. Chryso-obtusin (Figure 5B), which in the NAD-free structure localized near the adenine-binding region of the NAD cofactor (Supplementary Figures S1 A,B), adopted a markedly different orientation in the NAD-bound state due to steric clashes with the adenine moiety.
This positional shift suggests that Chryso-obtusin acts as a cofactor-competitive inhibitor, obstructing NAD association with the enzyme. In contrast, Episesamin, Norhyoscine, and Apohyoscine (Figures 5C-E) maintained consistent binding within the substrate-binding pocket in both NAD-free and NAD-bound conformations, as also supported by Supplementary Figures S1 C-H. These phytochemicals engaged the same catalytic residues as INH, indicating a substrate-competitive inhibition mechanism that does not disrupt NAD association. Together, these findings highlight two distinct inhibitory modes: stable substrate-competitive binding by Episesamin, Norhyoscine, and Apohyoscine, and dual interference with both substrate and cofactor binding by Chryso-obtusin-an approach that may offer advantages against drug-resistant Mtb. The superimposition of the docking poses all the compounds within the binding pocket of InhA target protein is presented in Supplementary Figure S2.
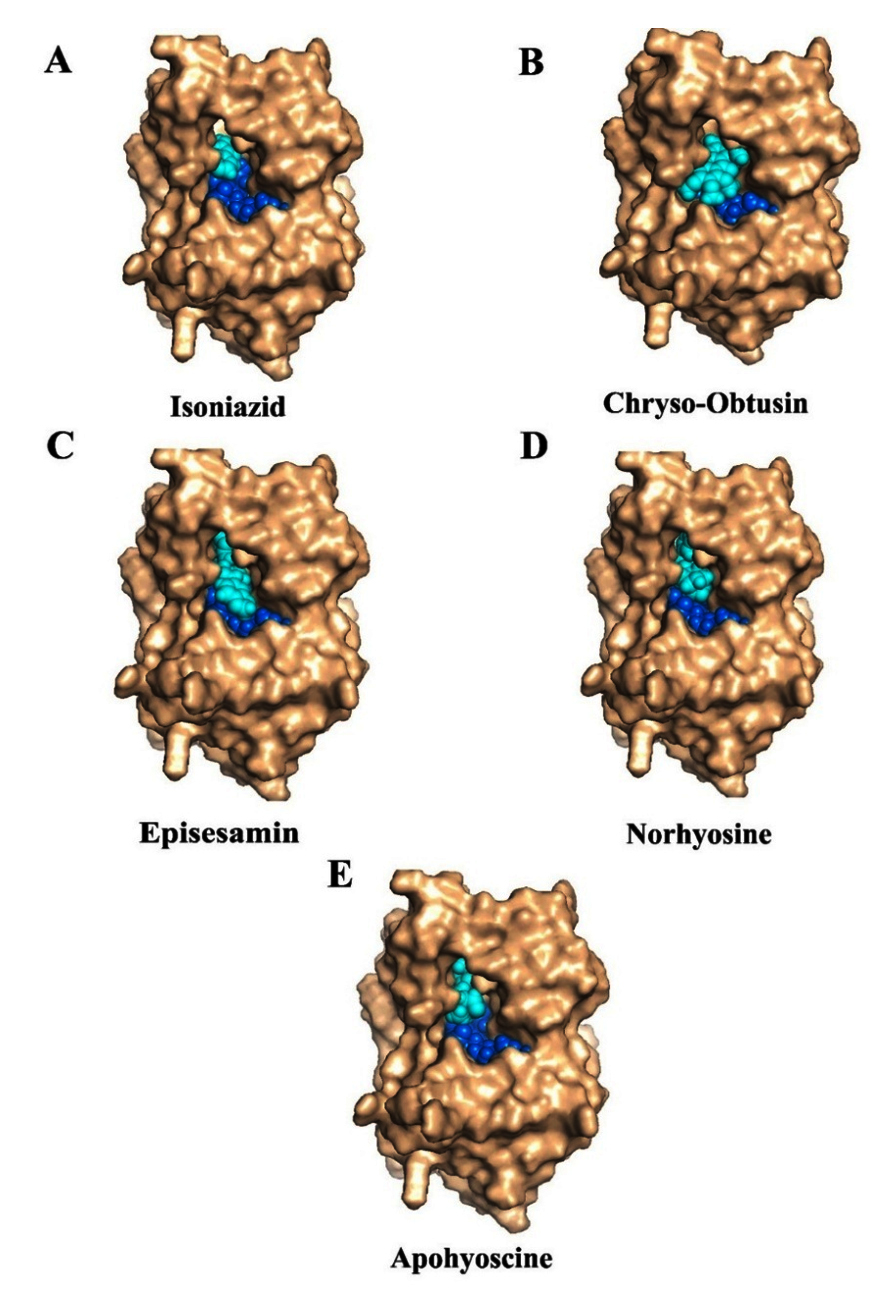
Figure 5. Molecular docking studies of herbal compounds on InhA-NAD complex: Docking of compounds (cyan color) on InhA-NAD (blue color) complex (A) Isoniazid, (B) Apohyoscine, (C) Chryso-obtusin, (D) Episesamin and (E) Norhyosine. These natural compounds target distinct cofactor binding sites and bind in parallel with NAD
Molecular dynamics simulation
To evaluate the structural stability of the ligand-InhA complexes, molecular dynamics (MD) simulations were performed using the Flare MD tool, integrated within the Cresset Flare V 6.0 software suite.40,41 The results of these simulations are presented as root-mean-square deviation (RMSD) and root-mean-square fluctuation (RMSF) plots (Figure 6).
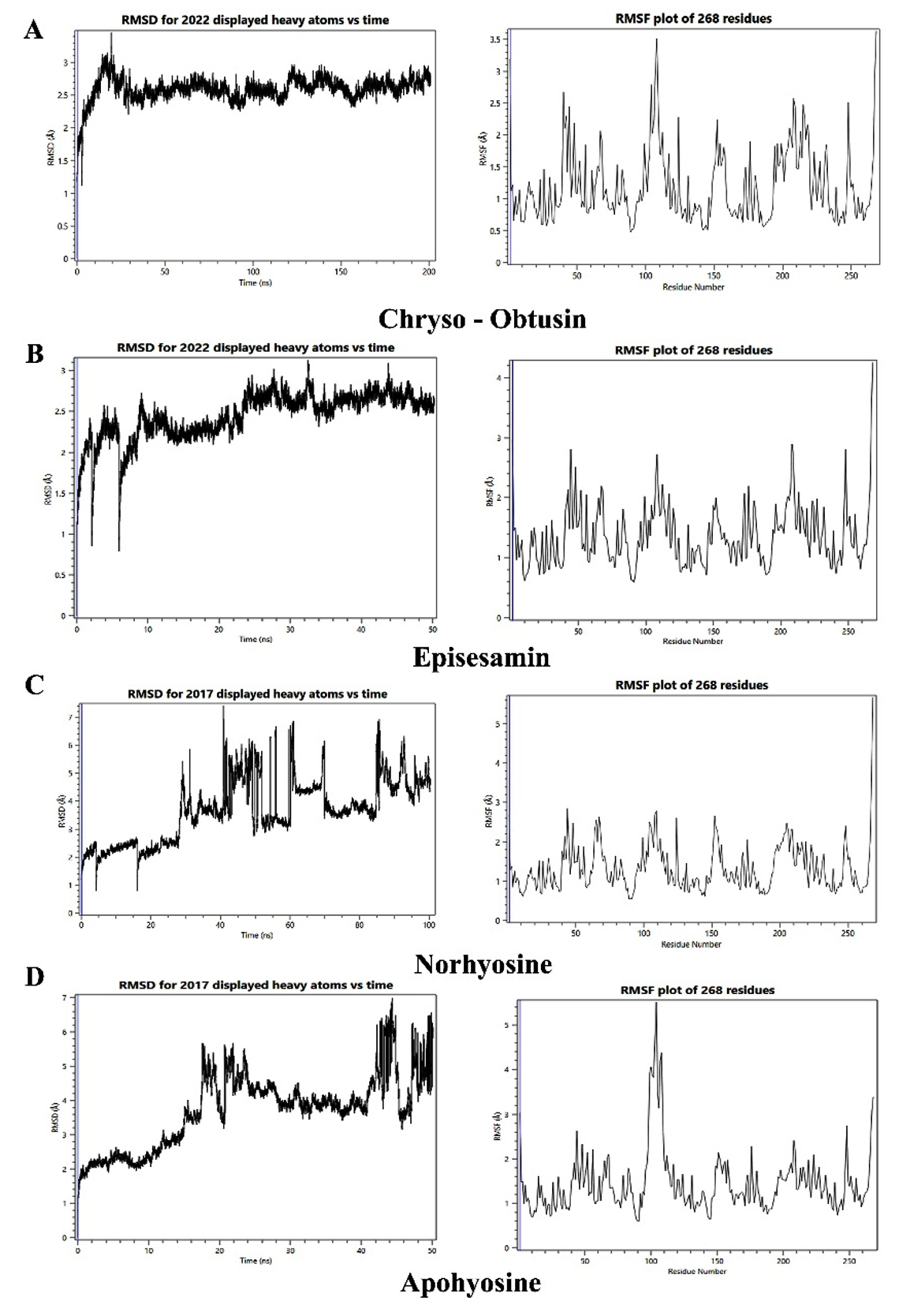
Figure 6. Molecular dynamics simulation analysis of InhA-ligand complexes: Root mean square deviation (RMSD, left panels) and root mean square fluctuation (RMSF, right panels) plots for InhA in complex with four phytochemicals: (A) Chryso-obtusin, (B) Episesamin, (C) Norhyoscine, and (D) Apohyoscine. RMSD analysis indicates that Chryso-Obtusin, Episesamin, and Norhyoscine maintained stable binding over the simulation period, with RMSD values largely within the acceptable range (<2.5 Å). In contrast, the Apohyoscine-InhA complex exhibited significant fluctuations, with RMSD exceeding 3.5 Å towards the end of the simulation, indicating reduced structural stability. RMSF plots reveal moderate residue-level fluctuations across all complexes, suggesting that ligand binding did not induce major conformational changes in the InhA protein. However, slightly elevated residue mobility is observed in the Apohyoscine-bound structure, supporting its reduced binding stability
Initially, all docked complexes were assessed for RMSD fluctuations of both the ligand and the protein, using the initial conformation as a reference frame. The RMSD values for InhA remained below 2.5 Å in most complexes, including Chryso-obtusin-InhA, Norhyoscine-InhA, and Episesamin-InhA, suggesting that the enzyme retained its structural integrity upon ligand binding without undergoing significant conformational perturbations. Notably, all ligand-protein complexes exhibited acceptable RMSD values, indicative of structural stability. However, the Apohyoscine-InhA complex displayed RMSD deviations exceeding 2.5 Å, suggesting reduced structural stability and potential conformational instability in this interaction.
To further investigate local structural variations within the ligand-protein complexes, RMSF measurements were employed, allowing for the detection of localized fluctuations at the atomic level. Analysis of RMSF values indicated that variations in amino acid residues of InhA, as well as atomic fluctuations within the ligand molecules, contributed to minor local structural changes in the docked complexes. Importantly, the RMSF values for InhA residues remained within an acceptable range, further supporting the structural stability of the ligand-bound enzyme.
Recent advances in anti-tuberculosis drug discovery have led to the identification of several Mtb proteins as promising therapeutic targets. Among these, enzymes involved in the biosynthesis of mycolic acids-an essential component of the Mtb cell wall-have been extensively investigated. Numerous inhibitors targeting this pathway, including isoniazid, triclosan, ethionamide, and pyrrolidine carboxamides have been reported to exert potent inhibitory effects by selectively targeting enzymes within the fatty acid synthase II (FAS-II) system.34,42
Although the Himalayan region is home to a vast array of medicinal herbs, only a select subset has been traditionally incorporated into Hawan samagri, a sacred polyherbal mixture used in Indian ethnomedicine for its therapeutic smoke. In this study, we focused on 27 of the most prominent Himalayan herbs that have been consistently reported for their use in Hawan practices over several decades. These selected herbs are well-documented in the literature for their richness in phytochemicals with diverse medicinal properties, including established antimicrobial activities.
From this ethnopharmacologically validated pool, we curated 37 previously unreported phytochemicals that, despite being present in bioactive plants, remain unexplored for their specific biological functions. These phytochemicals were subjected to molecular docking studies against Mtb InhA enzyme, a key target in the mycolic acid biosynthesis pathway. Molecular docking studies revealed that these phytochemicals preferentially occupied critical substrate-binding sites on InhA, with binding energies ranging from -8.74 to -9.82 kcal/mol. Chryso-obtusin emerged as the most promising compound, binding near the NAD cofactor-binding region, particularly the adenine moiety pocket. This suggests a cofactor-competitive mode of inhibition, where Chryso-obtusin may block NAD binding, thereby disrupting the enzymatic activity without competing directly with the substrate. In contrast, Episesamin, Norhyoscine, and Apohyoscine were found to localize within the highly conserved substrate-binding site, forming strong interactions with catalytically relevant residues such as Tyr158, Phe149, Met199, Leu218, and Thr196. These residues are also involved in binding the INH-NAD adduct, indicating that these compounds likely inhibit InhA through a substrate-competitive mechanism similar to INH. This dual-inhibition pattern suggests a potential strategy to overcome resistance mechanisms, particularly those linked to katG mutations that impair INH activation.
Each of these phytochemicals, with the exception of Apohyoscine, has been previously documented for its medicinal properties. For instance, Chryso-obtusin is known for its anti-cancer, anti-Alzheimer’s, anti-diabetic, platelet anti-aggregatory, and anti-Parkinson’s disease activities.43 Episesamin has demonstrated anti-neoplastic properties and potential therapeutic effects against obesity, while Norhyoscine has been reported for its efficacy in treating cancer, nausea, vomiting, and motion sickness.44,45 Notably, Apohyoscine lacks prior documentation regarding its pharmacological activity, thereby warranting further investigation as a novel bioactive compound.
MD simulations provided further insights into the stability of these ligand-protein complexes over time. RMSD analysis indicated that most phytochemical-InhA complexes remained stable throughout the simulation period, with minimal deviations observed in backbone conformations. The exception was the Apohyoscine-InhA complex, which exhibited an RMSD value exceeding 2.5 Å, suggesting a lower binding stability. RMSF analysis further corroborated these findings, showing acceptable fluctuations in amino acid residues, thereby affirming the structural integrity of the protein upon ligand binding. Importantly, the phytochemical ligands did not induce significant conformational changes in InhA, reinforcing the notion that these molecules act as direct inhibitors rather than allosteric modulators.
The distinct binding patterns of Chryso-obtusin and the remaining three phytochemicals highlight their potential for complementary inhibition mechanisms. Chryso-obtusin, by targeting the NAD-binding pocket, may serve as a novel inhibitor disrupting cofactor interactions, while Episesamin, Norhyoscine, and Apohyoscine mimic the mechanism of INH by obstructing substrate access at site 2. This dual-inhibition strategy may prove advantageous in overcoming drug-resistance, particularly in Mtb strains harboring katG mutations, which hinder the activation of INH. The absence of direct evidence linking these phytochemicals to anti-TB activity warrants further in vivo validation studies to confirm their efficacy in inhibiting mycolic acid biosynthesis.
Given the generally lower toxicity of plant-derived compounds, these phytochemicals offer promising leads for drug development. Overall, the distinct binding modes observed in this study suggest that phytochemicals from Himalayan herbs can act as novel InhA inhibitors through cofactor-competitive and substrate-competitive mechanisms. These findings underscore the need for further in vitro and in vivo validation, structure-activity relationship (SAR) analysis, and pharmacokinetic evaluation to optimize their therapeutic potential and establish their role in combating drug-resistant TB.
The findings of this study underscore the potential of Himalayan herb derived phytochemicals as novel InhA inhibitors. The identification of distinct binding modes, supported by molecular docking and MD simulations, suggests that these compounds may offer new therapeutic avenues for combating TB. However, in vitro enzyme inhibition assays and in vivo studies are essential to establish their true efficacy and therapeutic potential. Future investigations should focus on structure-activity relationship (SAR) studies, bioavailability assessments, and combination therapies to optimize these compounds for clinical application against drug-resistant Mtb strains.
This study highlights the promising potential of phytochemicals derived from Himalayan herbs traditionally used in Hawan samagri as novel therapeutic agents against Mtb. Using a combination of molecular docking and molecular dynamics simulations, we identified four lead compounds-Chryso-obtusin, Episesamin, Norhyoscine, and Apohyoscine-that exhibited high binding affinity and favorable interaction profiles with the InhA enzyme, a crucial component of the mycolic acid biosynthesis pathway. These compounds were not only found to engage key catalytic and cofactor-binding residues but also maintained structural stability within the enzyme’s active site throughout MD simulations, supporting their potential inhibitory efficacy.
Chryso-obtusin, in particular, showed a unique interaction with the cofactor-binding site of InhA, suggesting an alternative inhibitory mechanism distinct from that of conventional drugs like INH. Meanwhile, Episesamin, Norhyoscine, and Apohyoscine mimicked isoniazid’s mechanism by binding at the substrate-binding site, potentially blocking NADH access and enzymatic activity. The dual targeting of both the cofactor and substrate-binding sites by these phytochemicals presents a compelling strategy for overcoming current drug-resistance mechanisms in TB therapy.
Additional file: Additional Table S1-S4 and Figure S1-S2.
ACKNOWLEDGMENTS
The authors are grateful for the support from the members of the Research Committee, Department of Biological Sciences and Biotechnology, Chhatrapati Shahu Ji Maharaj University, Kanpur, Uttar Pradesh, India, for their constructive feedback and thoughtful suggestions that greatly enhanced the depth of this study.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
AUTHORS’ CONTRIBUTION
JS, DKV and RS conceptualized the study. DKV performed supervision. JS, DKV, and RS applied methodology. JS, VV, DKV and RS performed investigation. JS and DKV performed data curation and visualization. JS and DKV wrote original draft. JS and DKV wrote, reviewed and edited the manuscript. All authors read and approved the final manuscript for publication.
FUNDING
This research was funded by Chhatrapati Shahu Ji Maharaj University, Kanpur, and the Anusandhan National Research Foundation (ANRF) under the Prime Minister’s Early Career Research Grant Scheme, through the Large Research Group Project (Grant/File No.: ANRF/ECRG/2024/006549/LS).
DATA AVAILABILITY
All datasets generated or analyzed during this study are included in the manuscript and/or in the supplementary files.
ETHICS STATEMENT
Not applicable.
- Gill CM, Dolan L, Piggott LM, McLaughlin AM. New developments in tuberculosis diagnosis and treatment. Breathe (Sheff). 2022;18(1):210149.
Crossref - Seung KJ, Keshavjee S, Rich ML. Multidrug-Resistant Tuberculosis and Extensively Drug-Resistant Tuberculosis. Cold Spring Harb Perspect Med. 2015;5(9):a017863.
Crossref - Jankute M, Cox JA, Harrison J, Besra GS. Assembly of the Mycobacterial Cell Wall. Annu Rev Microbiol. 2015;69:405-23.
Crossref - Dulberger CL, Rubin EJ, Boutte CC. The mycobacterial cell envelope – a moving target. Nat Rev Microbiol. 2020;18(1):47-59.
Crossref - Brennan PJ, Nikaido H. The envelope of mycobacteria. Annu Rev Biochem. 1995;64:29-63.
Crossref - Alderwick LJ, Harrison J, Lloyd GS, Birch HL. The Mycobacterial Cell Wall—Peptidoglycan and Arabinogalactan. Cold Spring Harb Perspect Med. 2015;5(8):a021113.
Crossref - Francia MV, Weaver KE, Goicoechea P, Tille P, Clewell DB. Characterization of an active partition system for the Enterococcus faecalis pheromone-responding plasmid pAD1. J Bacteriol. 2007;189(23):8546-55.
Crossref - PaweLczyk J, Kremer L. The Molecular Genetics of Mycolic Acid Biosynthesis. Microbiol Spectr. 2014;2(4):MGM2-0003-2013.
Crossref - Faion L, Djaout K, Frita R, et al. Discovery of the first Mycobacterium tuberculosis MabA (FabG1) inhibitors through a fragment-based screening. Eur J Med Chem. 2020;200:112440.
Crossref - Duan X, Xiang X, Xie J. Crucial components of Mycobacterium type II fatty acid biosynthesis (Fas-II) and their inhibitors. FEMS Microbiol Lett. 2014;360(2):87-99.
Crossref - Vilcheze C, Jacobs WR, Jr. The mechanism of isoniazid killing: clarity through the scope of genetics. Annu Rev Microbiol. 2007;61:35-50.
Crossref - Vilcheze C, Jacobs WR, Jr. Resistance to Isoniazid and Ethionamide in Mycobacterium tuberculosis: Genes, Mutations, and Causalities. Microbiol Spectr. 2014;2(4):MGM2-0014-2013.
Crossref - Manjunatha UH, Rao SPS, Kondreddi RR, et al. Direct inhibitors of InhA are active against Mycobacterium tuberculosis. Sci Transl Med. 2015;7(269):269ra3.
Crossref - Jayabal D, Jayanthi S, Thirumalaisamy R, Karthika R, Iqbal MN. Comparative anti-Diabetic potential of phytocompounds from Dr. Duke’s phytochemical and ethnobotanical database and standard antidiabetic drugs against diabetes hyperglycemic target proteins: an in silico validation. J Biomol Struct Dyn. 2023;41(24):15137-15149.
Crossref - Banerjee A, Dubnau E, Quemard A, et al. inhA, a gene encoding a target for isoniazid and ethionamide in Mycobacterium tuberculosis. Science. 1994;263(5144):227-230.
Crossref - Gurib-Fakim A. Medicinal plants: traditions of yesterday and drugs of tomorrow. Mol Aspects Med. 2006;27(1):1-93.
Crossref - Raghuvanshi M, Pandya P, Joshi R. Yagyopathic Herbal Treatment of Pulmonary Tuberculosis Symptoms: A Clinical Trial. Alternative and Complementary Therapies. 2004;10:101-105.
Crossref - Rastogi V, Jyoti T, Patni T, Vijay C, Sharma P. Anti tubercular minimum inhibitory concentration (MIC) and chemical characterization of ethnobotanical mixture used in the treatment of tuberculosis. Indian J Microbiol Res. 2019;6(1):50-56.
Crossref - Ratnakaram V. Charaka Samhita Vol 1 (Sutra Sthan)- R. Vidyanath (1). 2020.
- Nautiyal CS, Chauhan PS, Nene YL. Medicinal smoke reduces airborne bacteria. J Ethnopharmacol. 2007;114(3):446-451.
Crossref - Fatwati K, Amin A, Indriani L, Ladju RB, Akbar FH, Hamrun N. GC-MS analysis and in silico approaches to Stichopus hermanii as anti-inflammatory through PKC-b inhibition. Results in Chemistry. 2025;14(15):102086.
Crossref - Lamie PF, Philoppes JN. Design, synthesis, stereochemical determination, molecular docking study, in silico pre-ADMET prediction and anti-proliferative activities of indole-pyrimidine derivatives as Mcl-1 inhibitors. Bioorg Chem. 2021;116:105335.
Crossref - Rozwarski DA, Vilcheze C, Sugantino M, Bittman R, Sacchettini JC. Crystal structure of the Mycobacterium tuberculosis enoyl-ACP reductase, InhA, in complex with NAD+ and a C16 fatty acyl substrate. J Biol Chem. 1999;274(22):15582-15589.
Crossref - Forli S, Huey R, Pique ME, Sanner MF, Goodsell DS, Olson AJ. Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat Protoc. 2016;11(5):905-919.
Crossref - Morris GM, Goodsell DS, Huey R, Olson AJ. Distributed automated docking of flexible ligands to proteins: parallel applications of AutoDock 2.4. J Comput Aided Mol Des. 1996;10(4):293-304.
Crossref - Sharma A, Islam MH, Fatima N, et al. Elucidation of marine fungi derived anthraquinones as mycobacterial mycolic acid synthesis inhibitors: an in silico approach. Mol Biol Rep. 2019;46(2):1715-1725.
Crossref - Morris GM, Goodsell DS, Halliday RS, et al. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem. 1998;19(14):1639-1662.
Crossref - Nakayama Y, Kaneko H. Development of New Molecular Descriptors Based on Flare Software Considering Three-Dimensional Chemical Structures. Ind Eng Chem Res. 2023;63(1):49-55.
Crossref - Umar HI, Ajayi A, Bello RO, et al. Novel Molecules derived from 3-O-(6-galloylglucoside) inhibit Main Protease of SARS-CoV 2 In Silico. Chem Zvesti. 2022;76(2):785-796.
Crossref - Rajasekhar S, Karuppasamy R, Chanda K. Exploration of potential inhibitors for tuberculosis via structure-based drug design, molecular docking, and molecular dynamics simulation studies. J Comput Chem. 2021;42(24):1736-1749.
Crossref - Anand U, Jacobo-Herrera N, Altemimi A, Lakhssassi N. A Comprehensive Review on Medicinal Plants as Antimicrobial Therapeutics: Potential Avenues of Biocompatible Drug Discovery. Metabolites. 2019;9(11).
Crossref - Awadelkareem AM, Al-Shammari E, Elkhalifa AEO, et al. Phytochemical and In Silico ADME/Tox Analysis of Eruca sativa Extract with Antioxidant, Antibacterial and Anticancer Potential against Caco-2 and HCT-116 Colorectal Carcinoma Cell Lines. Molecules. 2022;27(4).
Crossref - Takayama K, Wang C, Besra GS. Pathway to synthesis and processing of mycolic acids in Mycobacterium tuberculosis. Clin Microbiol Rev. 2005;18(1):81-101.
Crossref - North EJ, Jackson M, Lee RE. New approaches to target the mycolic acid biosynthesis pathway for the development of tuberculosis therapeutics. Curr Pharm Des. 2014;20(27):4357-78.
Crossref - Chollet A, Mourey L, Lherbet C, et al. Crystal structure of the enoyl-ACP reductase of Mycobacterium tuberculosis (InhA) in the apo-form and in complex with the active metabolite of isoniazid pre-formed by a biomimetic approach. J Struct Biol. 2015;190(3):328-337.
Crossref - Geng S, Sang X, Liu X, Ren Y, Xue Y. A theoretical study of UV-Vis spectrum and antioxidant activity of chryso-obtusin. J Theor Comput Chem. 2018;17(02):1850015.
Crossref - Garang Z, Feng Q, Luo R, et al. Commiphora mukul (Hook. ex Stocks) Engl.: Historical records, application rules, phytochemistry, pharmacology, clinical research, and adverse reaction. J Ethnopharmacol. 2023;317:116717.
Crossref - Murugan P, Salim A, Khusro A, et al. Anti-pathogenic, anti-diabetic, anti-inflammatory, antioxidant, and wound healing efficacy of Datura metel L. leaves. Arabian Journal of Chemistry. 2022;15(1):104112.
Crossref - Akbar S. Handbook of 200 Medicinal Plants: A Comprehensive Review of Their Traditional Medical Uses and Scientific Justifications. 2020. Switzerland, Springer Cham
Crossref - Samways ML, Macdonald HEB, Essex JW. grand: A Python Module for Grand Canonical Water Sampling in OpenMM. J Chem Inf Model. 2020;60(10):4436-4441.
Crossref - Melling OJ, Samways ML, Ge Y, Mobley DL, Essex JW. Enhanced Grand Canonical Sampling of Occluded Water Sites Using Nonequilibrium Candidate Monte Carlo. J Chem Theory Comput. 2023;19(3):1050-1062.
Crossref - Prasad MS, Bhole RP, Khedekar PB, Chikhale RV. Mycobacterium enoyl acyl carrier protein reductase (InhA): A key target for antitubercular drug discovery. Bioorg Chem. 2021;115:105242.
Crossref - Ali MY, Park S, Chang M. Phytochemistry, Ethnopharmacological Uses, Biological Activities, and Therapeutic Applications of Cassia obtusifolia L.: A Comprehensive Review. Molecules. 2021;26(20):6252.
Crossref - Freise C, Trowitzsch-Kienast W, Ruehl M, et al. (+)-Episesamin exerts anti-neoplastic effects in human hepatocellular carcinoma cell lines via suppression of nuclear factor-kappa B and inhibition of MMP-9. Invest New Drugs. 2012;30(6):2087-95.
Crossref - Freise C, Trowitzsch-Kienast W, Erben U, et al. (+)-Episesamin inhibits adipogenesis and exerts anti-inflammatory effects in 3T3-L1 (pre)adipocytes by sustained Wnt signaling, down-regulation of PPARg and induction of iNOS. J Nutr Biochem. 2013;24(3):550-555.
Crossref
© The Author(s) 2025. Open Access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License which permits unrestricted use, sharing, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.