


ISSN: 0973-7510
E-ISSN: 2581-690X
Helicobacter pylori’s type IV secretion system (T4SS) is a key virulence determinant, mediating the delivery of effector proteins such as cytotoxin-associated gene A (CagA) into host epithelial cells. Despite its significance, the molecular architecture and assembly dynamics of the inner membrane core complex within the Cag-T4SS remain inadequately resolved. This study aimed to identify and characterize the direct interaction between CagF, a chaperone protein, and CagV, an inner membrane-associated component of the Cag-T4SS. Immunoprecipitation and pull-down assays confirm the physical interaction between CagF and CagV. Computational docking and molecular dynamics simulations further elucidated the binding interface, revealing a stable interaction mediated by electrostatic complementarity, hydrogen bonding, and salt bridges. Co-expression and pull-down experiments using recombinant proteins validated these findings. Notably, deletion analysis identified the critical role of the N-terminal region (residues 50-100) of CagF in CagV binding, further supporting the computational predictions. Given the essential role of CagV in CagA translocation, we hypothesize that CagF-CagV interaction may facilitate the localization of the CagF-CagA complex near the secretion channel, promoting efficient effector translocation. These findings enhance our understanding of the Cag-secretion system’s structural organization and biogenesis.
H. pylori, Cag-type IV Secretion System, CagA, CagF, CagV
The World Health Organization declared Helicobacter pylori (H. pylori) a class I carcinogen.1 While often asymptomatic, it can persist in the gastric lumen for life and cause gastric ulcers, mucosa-associated lymphoid tissue lymphoma, and gastric adenocarcinoma. It encodes a cag pathogenicity island (cag-PAI), comprising 27 Cag proteins, including virulence factor cytotoxin-associated gene A (CagA). These proteins constitute a syringe-like Cag-type IV secretion system (Cag-T4SS),2 which translocates CagA into gastric cells in a contact-dependent manner.3 Translocated CagA perturbs multiple host signaling pathways, resulting in dysregulated cell proliferation, altered adhesion and morphology, and enhanced pro-inflammatory responses, which together promote gastric carcinogenesis.4,5 Additionally, H. pylori promote gastric carcinogenesis by persistently activating multiple oncogenic pathways (such as STAT3, NF-kB, Akt, and Wnt/β-catenin), inducing inflammation-driven DNA damage and mutations, and disturbing cell polarity and motility. The chronic inflammatory response it elicits is a key driver of this malignant transformation.6 A recent study investigating the genotyping of H. pylori CagA and CagE strains in gastric mucosal samples has demonstrated a positive correlation between the presence of these genotypes and the severity of gastric lesions in infected patients.7 This association was particularly pronounced in specific histopathological subtypes, including atrophic gastritis and intestinal metaplasia, suggesting a potential role of these virulence factors in the progression of gastric mucosal damage. Studies on systematic mutagenesis have identified 17 essential genes for CagA translocation, with others influencing its efficiency.2 The overarching architecture of the Cag-T4SS comprises an outer membrane core complex (OMCC), a cytosolic/inner membrane core complex (IMCC), and a pilus-like structure.8 Cryo-electron tomography and crystallographic studies of Cag-T4SS have revealed that CagX, CagY, CagM, CagT, and Cag3 are essential structural components of the OMCC of the secretion system.9,10 The OMCC of Cag-T4SS has been more extensively studied owing to the significant homology of its proteins with those of prototypical T4SS, such as Agrobacterium tumefaciens (A. tumefaciens). In contrast, the IMCC remains less characterized, despite its critical role in the assembly and function of the Cag-T4SS. The IMCC comprises several key components, including ATPases (CagE, Cagα, and Cagβ), integral membrane proteins (CagW and CagV), and accessory inner membrane-anchored proteins such as CagF, CagZ, and CagU, all of which are essential for CagA translocation and the biogenesis of the secretion system.8,11-13 The precise molecular interactions, spatial organization, and individual contributions of these proteins to the stability and functional integrity of the IMCC remain largely uncharacterized.
The interaction between CagA and CagF has been demonstrated both with recombinant proteins and under in vivo conditions.14 This interaction operates independently of other Cag-T4SS components and occurs before the constitution of the secretion channel.15 Although CagF lacks a membrane-localizing domain, cell fractionation experiments show its presence in the membrane pool, probably owing to interactions with other Cag membrane proteins. In contrast, CagV, with a transmembrane domain, has been exclusively detected in the membrane fraction.16 The interaction between CagF and CagA is hypothesized to play a critical role in the early stages of the CagA translocation process. CagF appears to stabilize CagA upon its synthesis, potentially preventing its degradation into small fragments of 100 and 35 kDa. However, CagF disengages from CagA at a specific step during the translocation process, as it is not detected at the pilus tip or within the host cell.15 These findings highlight the temporal and spatial specificity of the CagF-CagA interaction, emphasizing the need for further studies to uncover its mechanism and identify potential Cag partner(s) within the inner membrane complex. Previously, our laboratory has reported that recombinant CagF pulled down CagV, CagZ, and CagA from H. pylori cell extract via MBP pull-down assay.16 However, no further study was performed to investigate its direct interaction with other cag proteins or its specific role in recruiting CagA into the secretion channel. Investigating the CagF-CagV interaction could provide critical insights into the structural organization and biogenesis of IMCC of the Cag-secretion system.
Bacterial strains and their growth conditions
The wild-type (WT) H. pylori strain 26695 was cultured following established protocols.17 Escherichia coli strains DH5a and BL21 (DE3) were transformed with the respective plasmids for cloning and recombinant protein expression, and subsequently cultured in selective media containing the appropriate antibiotics.
Cloning and expression of cagF and cagV
The cagF and cagV genes were amplified from the WT H. pylori (26695) genomic DNA using gene-specific primers. The resulting amplicons were integrated into pGEX-6P-2 and pMAL-c2X vectors to express as N-terminal GST- and MBP-tagged fusion proteins, respectively. pACYC-Duet1 vectors were used to express CagV as an untagged protein. Deletion constructs of cagF, including N-terminal deletions spanning residues 1-50 (Δ50NCagF) and 1-100 (Δ100NCagF), as well as C-terminal deletions spanning residues 219-268 (CagFΔ49C) and 169-268 (CagFΔ99C), were generated (Table). Table provides the oligonucleotide sequences used to amplify the target genes. Transformed E. coli BL21(DE3) cells were grown and induced with isopropyl β-D-thiogalactoside (IPTG) at optimal conditions.16 Co-expression was performed using pACYC–cagV/pGEX–cagF plasmid pairs, with double antibiotic selection (chloramphenicol and ampicillin).18 The co-expression protocol mirrored single-gene expression, except for dual antibiotic supplementation in the culture media.
Table:
List of oligonucleotides
Oligos |
Sequences |
|---|---|
FcagF BamHI |
5′-CCGGATCCATGAAACAAAGTTTGCGC-3′ |
FΔ50NcagF BamHI |
5′-CCGGATCCAACCTTACCGCTCTTTAT-3′ |
FΔ100NcagF BamHI |
5′-CCGGATCCTCATTTCTCAGTAATCAG-3′ |
RcagFΔ49C SalI |
5′-ATGCGTCGACTTACAAATGCGTGAAGTCATA-3′ |
RcagFΔ99N SalI |
5′-ATGCGTCGACTTACATGAAGTAAACATAAAC-3′ |
RcagF SalI |
5′-ATGCGTCGACTCAATCGTTACTGTTGTT-3′ |
FcagV BamHI |
5′-CGGGATCCATGTTAGGGAAAAAAAACGA-3′ |
RcagV SalI |
5′-ATGCGTCGACTTATTTATTTAATGCCTTATTTTT-3′ |
SDS-PAGE and Western blotting
SDS-PAGE was employed as mentioned by Laemmli.19 Proteins were transferred to a PVDF membrane using a semi-dry transfer system (Bio-Rad). After blocking with 5% Bloto (Genotech) in TBST (40 mM Tris/HCl, pH 7.2, 100 mM NaCl, 0.2% Tween-20), membranes were incubated overnight at 4 °C with primary antibodies, followed by HRP-conjugated secondary antibodies (G-Biosciences). Immunoreactive bands were detected using enhanced chemiluminescence. CagV (rabbit) and CagF (mouse) antibodies were used at 1:10,000 and 1:5,000 dilutions, respectively.
Co-immunoprecipitation
Co-immunoprecipitation was performed using either recombinant protein extracts or H. pylori cell lysates. Cells were treated with dithiobis (succinimidyl propionate) (DSP) (G-Biosciences, MO, USA) to stabilize the transient interaction between the proteins. H. pylori cells (~50 µL pellet) were lysed to perform co-immunoprecipitation, following the published protocol.17 To solubilize membrane-associated proteins, n-Dodecyl-β-D-maltoside (DDM) was used, along with lysis buffer.
Immunofluorescence microscopy (IFM)
H. pylori cells were subjected to immunofluorescence staining following published protocols with necessary modifications.17,20 Primary antibodies, a-CagV and a-CagF, were used in a dilution of 1:500. Cells were then incubated with fluorescent secondary antibodies, i.e., Alexa Fluor 488-conjugated goat anti-rabbit IgG and Alexa Fluor 594-conjugated goat anti-mouse IgG (Molecular Probes) for 1 h at room temperature. After washing, cells were mounted in 20% glycerol and proceeded to capture images using fluorescence microscopy (Carl Zeiss). Images were analyzed using AxioVision 4.8 software.
GST pull-down assay
Cell pellets containing N-terminal GST-tagged proteins and their variants were resuspended in lysis buffer Tris-HCl, pH 8.0, 100 mM NaCl, 2 mM EDTA, 1 mM PMSF, 1% Triton X-100, 0.2% sodium deoxycholate, 2 mM DTT, 10% glycerol, and 0.2 mg/mL lysozyme, lysed by sonication, and centrifuged at 10,000 rpm for 20 min at 4 °C. The supernatant was diluted 1:1 with binding buffer (50 mM Tris-HCl, pH 7.2, 100 mM NaCl, 1% Triton X-100, 1 mM EDTA, 1 mM PMSF, and 6 µL of 100x protease inhibitor cocktail). CagV was soluble in the suspension and used as prey. Protein concentrations were estimated using the CB-X kit (G-Biosciences). GST-tagged proteins used as bait, were immobilized on Glutathione resin (Himedia, India). Subsequently, CagV extract was allowed to bind with Glutathione-bound GST-CagF and its deletion variants for 1 h at 4 °C. Beads were thoroughly washed, and bound proteins were eluted with SDS sample buffer and analyzed by western blotting using appropriate primary antibodies.
Structural prediction and minimization
Due to the lack of an experimentally determined CagF structure, homology modeling was employed using the Swiss-Model server, with J0BZ86.1. A as the template (97.39% sequence identity). An unidentified amino acid at position 264 in the CagF sequence was replaced with alanine. For CagV, the structure with PDB ID: 6IQT was used; the first 73 N-terminal residues were missing, and residues 25-75 were predicted to form a transmembrane helix (TMHMM server).21 As this region is membrane-embedded and unlikely to interact with cytosolic proteins, only residues 74-252 were modelled.22 Both structures were refined by 200 ns molecular dynamics (MD) simulations to improve structural stability.
Intermolecular docking of proteins
HADDOCK v.2.2 web server was employed to dock CagF with CagV.23,24 Since no interface information was available, solvent-accessible residues for CagF and CagV were identified using NAccess v2.1.1.25 Residues with relative accessibility >100 are considered active and potentially involved in binding interactions. Rigid body docking generated 1000 initial structures, of which 200 underwent semi-flexible refinement and explicit solvent refinement. Clustering was performed based on the fraction of common contacts (FCC),26 using a cut-off of 0.6 and a minimum cluster size of four. The best cluster was chosen based on a more negative HADDOCK score, and a low Z-score, indicating better binding energy and cluster homogeneity.27 The optimal docking pose was chosen from the top cluster using these criteria.
Calculation of electrostatic surface and protein-protein interface
The best docking pose was processed using PDB2PQR to assign the titration states using PROPKA at pH 7.0.28,29 Electrostatics calculations were executed by Adaptive Poisson-Boltzmann Solver (APBS),30 and the images were rendered in Chimera.31 The protein-protein interface was analyzed using the PDBsum web server.32
Molecular dynamics simulation
Molecular dynamics (MD) simulations were conducted using the GROMACS v.2024.1 suite.33-35 Systems comprising CagF, CagV, or their complex, were modelled with the CHARMM36 force field36,37 and water was represented using the TIP3P model.38 Each system was solvated in a dodecahedral box, maintaining at least 1.0 nm of spacing between the protein surface and the box edges. Periodic boundary conditions were employed, and long-range electrostatic interactions were computed using the particle mesh Ewald (PME) method, with a grid spacing of 0.12 nm and fourth-order B-spline interpolation. A cutoff of 1.2 nm was applied to both real-space electrostatic and van der Waals interactions. Bond constraints were maintained using the LINCS algorithm,39 and the equations of motion were integrated with a 2 fs time step. For the trajectory analysis, at an interval of every 10 ps, the system coordinates were recorded. Pressure coupling was performed with a Parrinello-Rahman barostat40 set to 1 bar, with a coupling constant of 2.0 ps and a compressibility of 4.5 × 10-5 bar, employing an isotropic scaling scheme. Counter-ions were added by replacing water molecules to neutralize the system. All initial structures underwent energy minimization and equilibration according to the CHARMM protocol. Production simulations were performed in triplicate for 200 ns at 300 K.
Estimation of binding free energies with residue-level decomposition profile
The intermolecular binding free energy of the CagF-CagV complex was evaluated via Molecular Mechanics/Poisson-Boltzmann Surface Area (MM/PBSA) approach.41,42 The binding free energy, ΔGbind, was calculated according to the following equation:
ΔGbind = Gcomplex – (GCagF + GCagV)
Per residue, energy decomposition was also performed to identify the contributions of individual residues to the binding energy.
Interaction between CagF and CagV in H. pylori
We confirmed the CagF-CagV interaction by co-immunoprecipitating endogenous CagF from H. pylori extract using anti-CagV antibody (Figure 1A). No protein band was observed in the negative control, where pre-immune serum was used. This experiment was repeated under similar conditions using an anti-CagF antibody, which did not show any interaction between the proteins. This may be due to the weak or transient nature of the CagF-CagV interaction or the inability of anti-CagF antibody to pull CagV, a multi-interacting protein of Cag-T4SS. When purified recombinant CagF (i.e., MBP-CagF) was incubated with WT H. pylori cell extract, anti-CagV antibody successfully immunoprecipitated MBP-CagF in the presence of native CagV. However, no protein bands were observed when MBP-CagF was used alone (Figure 1B). These results suggest a potential physical interaction between CagF and CagV. To further substantiate this interaction, we analyzed their co-localization within H. pylori cells via immunofluorescence microscopy. As shown in Figure 1C, CagV co-localized with CagF at distinct foci within the bacterial inner membrane, while no fluorescence signal was detected with pre-immune serum (data not shown), suggesting a physical interaction between the two proteins.
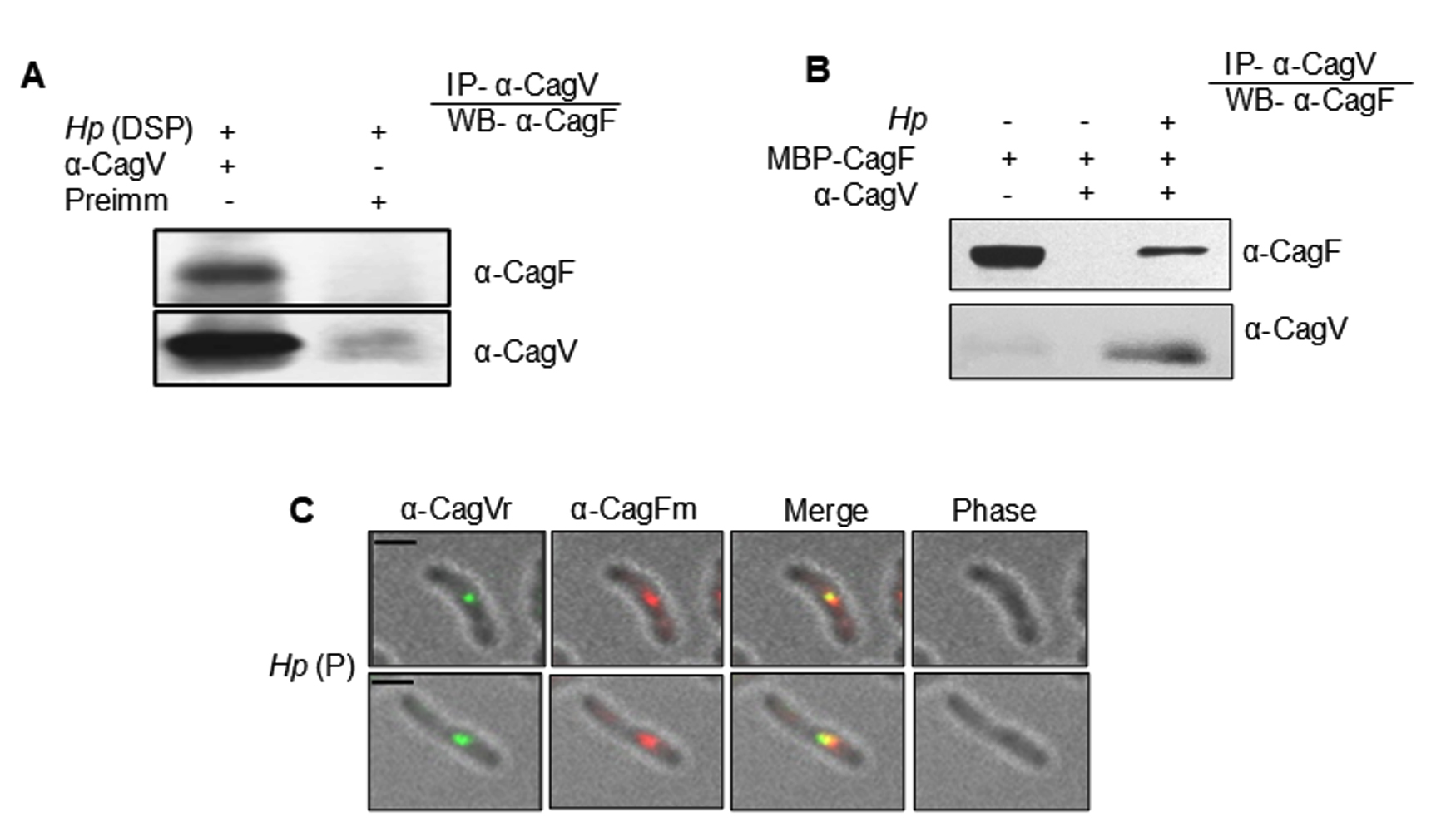
Figure 1. Co-immunoprecipitation and co-localization analyses show the interaction between CagF and CagV. (A) Western blot showing CagV antibody co-immunoprecipitating native CagF from H. pylori (Hp) extract. (B) Western blot showing co-immunoprecipitation of recombinant CagF (MBP-CagF) using CagV antibody from H. pylori extract. Pre-immunized serum (Preimm) was used as control. Dithiobis (succinimidyl propionate) (DSP) was used to stabilize the interaction between the proteins. (C) Immunofluorescence microscopy showing co-localization of CagV/CagF in permeabilized H. pylori cells, with Alexa Fluor 488 (green) and Alexa Fluor 594 (red) secondary antibodies. ‘r’ and ‘m’ denote rabbit and mouse primary antibodies, respectively; Scale bar = 2 µm
CagF interacts with CagV independently
To investigate the physical interaction between CagF and CagV, we employed computational docking and molecular dynamics simulations to identify binding interfaces, interaction stability, and key residues involved in the CagF-CagV complex formation. Notably, the structure of CagF has not been experimentally determined. Therefore, a structural model of CagF was generated through homology modeling using the SWISS-model server,43 with “J0BZ86.1. A” as a template. However, the crystal structure of CagV (PDB ID 6IQT) was utilized, as described in a previous study.22 Both structures were independently simulated to obtain stable conformations (Figure S1, S2), that were used as inputs for docking through HADDOCK.23,24 The resulting CagF-CagV complex was further simulated until it stabilized, providing deeper insights into the molecular nature of their interaction (Figure 2A). MD simulation analysis confirmed the stability and plausibility of the predicted docking interaction between CagF and CagV. This approach mirrors previous studies that used MD simulations to validate docking between Penaeus vannamei ferritin and Vibrio parahaemolyticus HrpE/YscL proteins, thus supporting the reliability of the current docking predictions.44 The surface localization of electrostatic charges on the CagF-CagV complex obtained through Advanced Poisson-Boltzmann Solver (APBS)30 revealed large clusters of negative charge throughout the CagF surface; however, the CagV surface was observed to be predominantly neutral with dispersed patches of positive charges (Figure 2B). This finding also aligns with the individual theoretical isoelectric points (pI) of CagF and CagV, 4.49 and 9.54, respectively, calculated through the Expasy tool.45 The distribution of opposing charges on the interface of the complex might have facilitated their binding with each other. The CagF-CagV complex gained conformational stability at approximately 125 ns as its root mean square deviation (RMSD) plateaued at 0.5 nm (Figure 2C). The radius of gyration (Rg) beyond 125 ns further confirmed that the complex is reasonably compact and stably folded (Figure 2D). The binding free energy (ΔGbind) was calculated using Molecular Mechanics Poisson-Boltzmann Surface Area (MMPBSA),41,42 yielding an average value of -49.58 ± 0.15 Kcal/mol (Figure 2E), which indicates spontaneous and energetically favourable complex formation. Hydrogen bonding (H-bond) is often considered crucial for stabilizing protein-protein complexes.46 Figure 2F shows the dynamic number of H-bonds formed in the CagF-CagV complex along a 200 ns trajectory. Between 100-200 ns, the average number of H-bonds stabilized around eight with transient drops to six and four at 110 and 140 ns, respectively, implying a stable interaction or minimal conformational change in the complex.
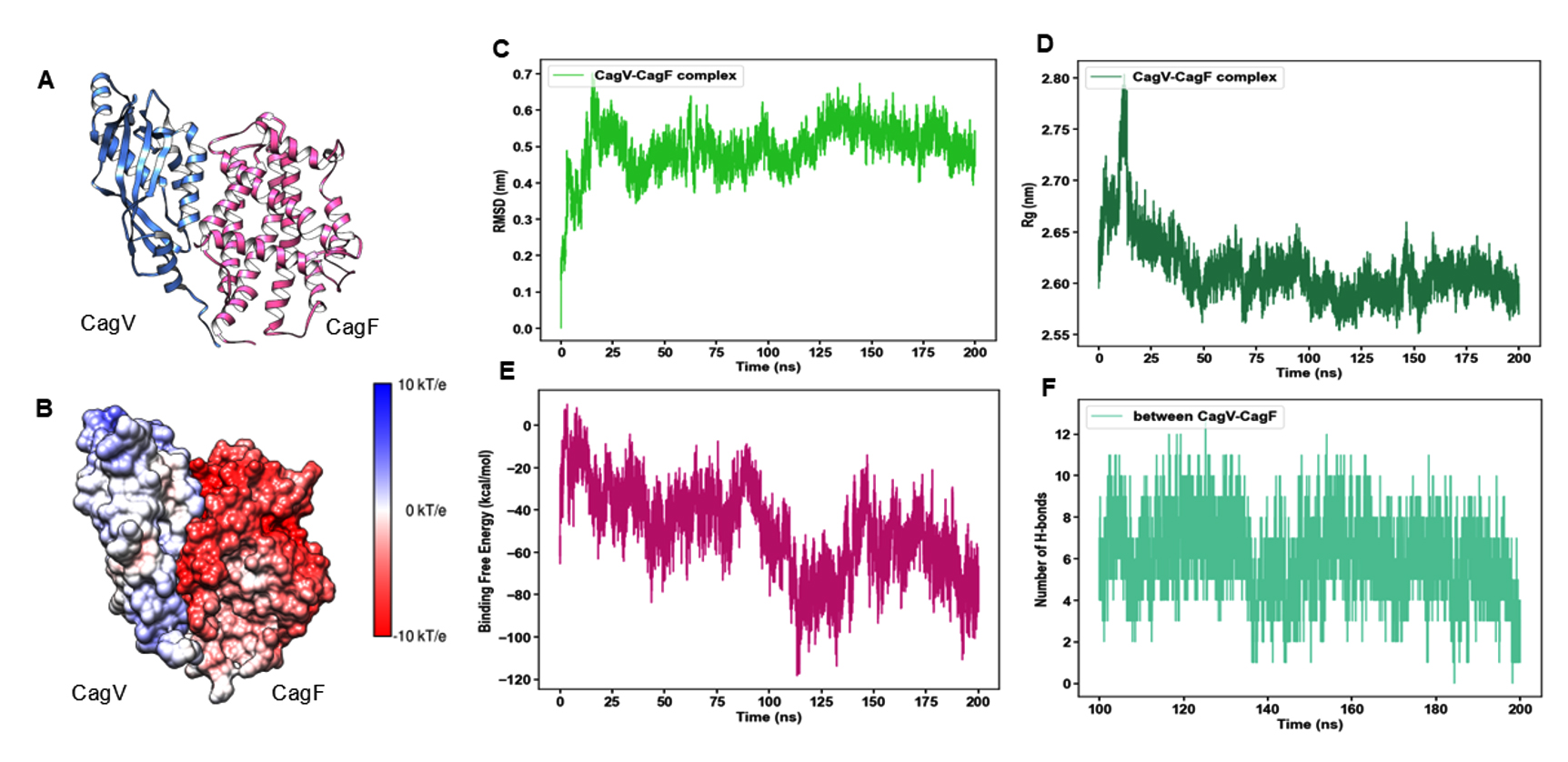
Figure 2. Molecular docking and MD simulation of CagF-CagV complex. (A) Cartoon representation of the CagF-CagV complex. (B) Surface representation of the electrostatic charge distribution at the interacting interface of the CagF-CagV complex. Colour scale: Blue, positively charged; red, negatively charged; and white, neutral surfaces. (C) Time dependence of Cα root mean square deviation (RMSD) of CagF-CagV complex. (D) Radius of gyration (Rg) of CagF-CagV complex against time. (E) Binding free energy of CagF-CagV complex over time. (F) Number of Hydrogen bonds (H-bonds) formed in 200 ns complete trajectory
To validate the in silico findings, protein-protein interaction assays were performed using recombinant proteins. MBP-CagF and untagged CagV (CagVA) were co-expressed in a heterologous E. coli system (Figure 3A). Co-expression facilitates correct folding and promotes stable complex formation in solution. Both proteins were recovered in the soluble fraction (data not shown). Immunoprecipitation of the co-expressed proteins using either anti-CagF or anti-CagV antibodies resulted in successful co-precipitation of their respective interaction partners (Figure 3B). However, no protein bands were detected when pre-immune serum was used as a negative control. These results strongly support a physical interaction between CagF and CagV, consistent with the in silico predictions.
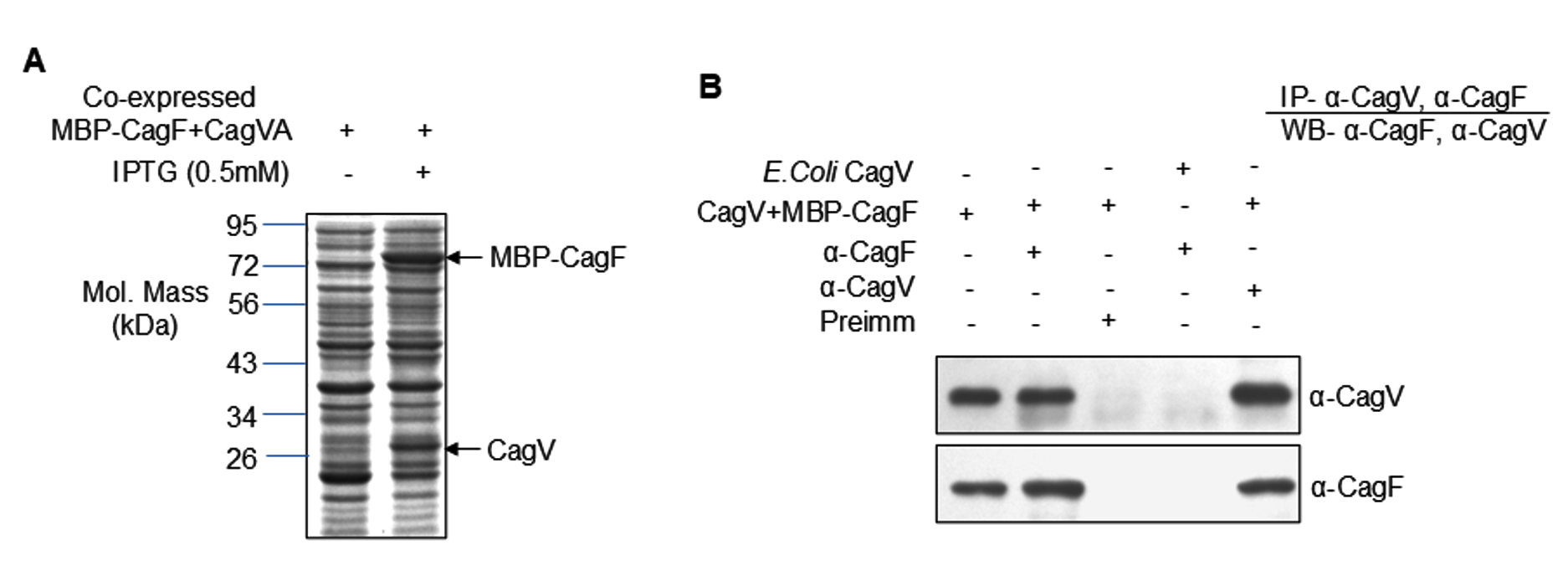
Figure 3. Expression and interaction analysis of recombinant CagVA and MBP-CagF. (A) SDS-PAGE analysis showing co-expression of recombinant CagVA (untagged) and MBP-CagF as indicated. The arrow marks the position of the recombinant proteins. The gel was stained with Coomassie Brilliant Blue. (B) Western blot analysis demonstrating co-immunoprecipitation of CagF and CagVA using anti-CagV and anti-CagF antibodies, respectively. Pre-immune (Preimm) serum served as a negative control. Antibodies used are indicated on the blots
The N-terminal region of CagF is essential for its interaction with CagV
Given the direct CagF-CagV interaction, we try to identify the minimal interaction region of CagF. MD simulations were performed to evaluate structural stability, binding affinity, and key interacting residues under physiologically relevant conditions. The MD simulation data revealed that the CagF-CagV interaction interface spans approximately 1,131 Å2, comprising 126 non-bonded contacts, 15 hydrogen bonds, and five salt bridges. Specifically, residues R114, R141, K146, K117, and R241 of CagV interacted with D57, D188, E71, E49, and E82 of CagF, respectively (Figure 4A and 4B). The structural orientation of the protein complex revealed that the N-terminal region of CagF (primarily residues 50-100) interacts with the C-terminal region of CagV, suggesting the presence of a defined interaction interface. Per-residue binding free energy decomposition identified several key residues in both CagF and CagV that significantly contribute to the entire complex’s cumulative binding free energy. Notably, all interfacial residues from both proteins contributed favorably to the stability of the complex. R238, R134, and K117 from CagV, along with L84 and Y56 from CagF, were the highest contributors. Specifically, R238 (CagV) formed a nonbonded contact with H83 (CagF), R134 (CagV) formed a H-bond with N187 (CagF), and K117 (CagV) formed both a H-bond and a salt bridge with E49 (CagF). Additionally, L84 and Y56 (CagF) were engaged in nonbonded interactions with V144 and N137 (CagV), respectively (Figure 4C). These findings highlight the crucial role of transient H-bonds and nonbonded interactions, alongside electrostatic salt bridges, in stabilizing the CagF-CagV complex.
These findings were validated biochemically by constructing and expressing a series of CagF deletion mutants (GST-Δ50NCagF, GST-Δ100NCagF, GST-CagFΔ49C, and GST-CagFΔ99C). SDS-PAGE confirmed their solubility (Figure S3A), while Western blotting showed partial degradation in all variants, irrespective of N- or C-terminal deletions (Figure S3B). These observations suggest that CagF may be inherently unstable in solution or require a binding partner for stability. To map the interaction region, GST pull-down assays were performed using CagF deletion variants as bait and CagVA as prey, with GST alone as a negative control. Western blot analysis revealed that CagV efficiently interacted with the C-terminal deletion mutants of CagF, whereas binding was significantly reduced in N-terminal deletion mutants, particularly Δ100NCagF (Figure 4D and 4E). However, no interaction was detected when GST alone was used. These results confirm that the N-terminal region of CagF is essential for its interaction with CagV, consistent with the computational predictions from MD simulations.
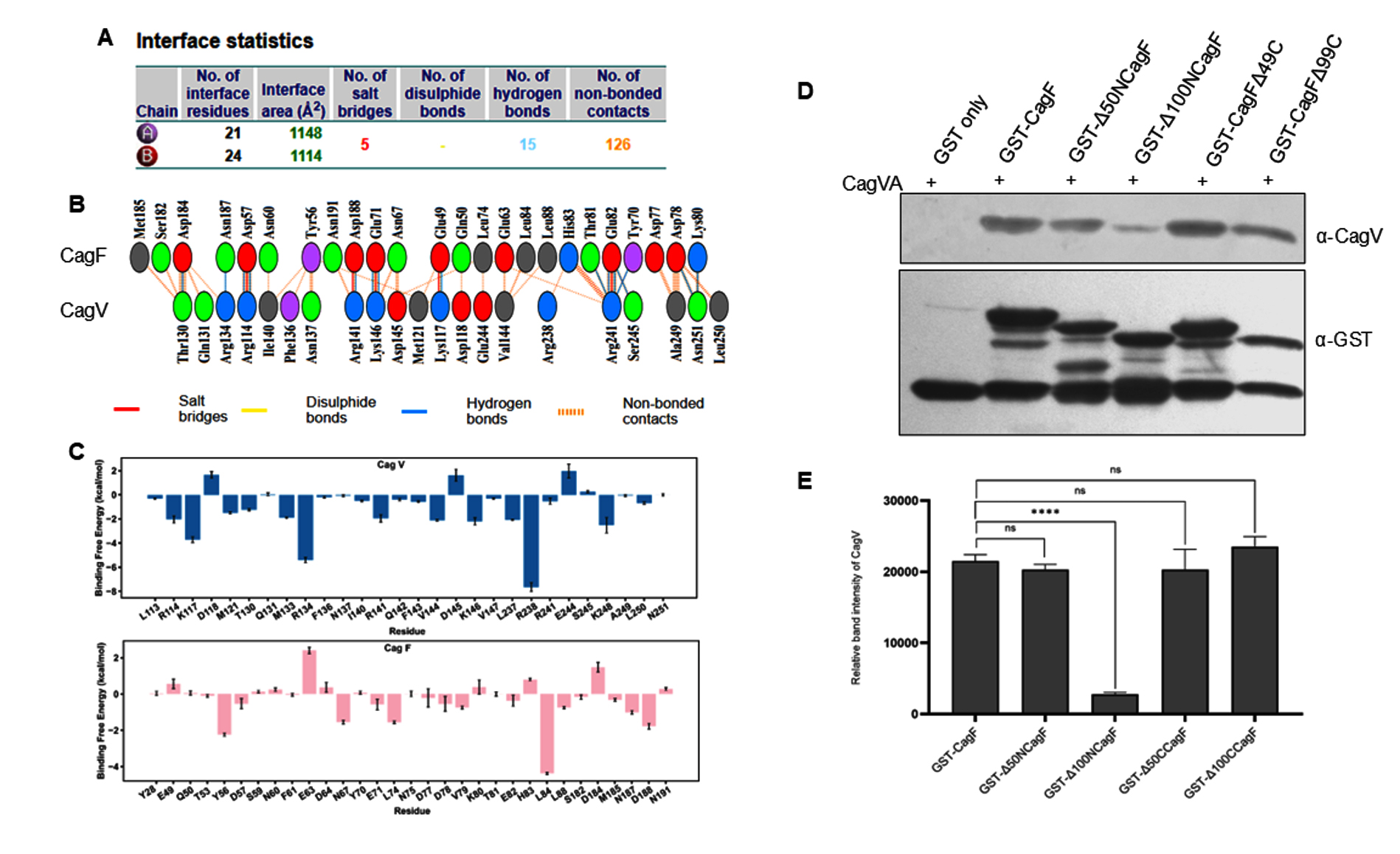
Figure 4. Interacting domain of CagF involved in stabilizing the CagF-CagV complex. (A) Interface statistics, with Chains A and B representing CagV and CagF, respectively. (B) visualization of the interacting residues at CagF-CagV interface. (C) Comparison of binding free energies of individual residues of CagV and CagF. (D) GST pull-down assay to show the interaction profile between recombinant CagVA (without tag) and the indicated CagF deletion mutant. (E) Quantification of band intensities from three independent experiments using IMAGEJ, presented as mean ± SD. Statistical analysis was done using Student’s t-test; **** indicates p < 0.0001
The H. pylori Cag-T4SS represents a uniquely complex, membrane-spanning delivery apparatus, specialized for the translocation of bacterial effector molecules such as CagA and peptidoglycan into host gastric epithelial cells. In contrast, the prototypical VirB/VirD4 T4SS of Agrobacterium tumefaciens is a more minimal system that primarily delivers T-DNA into plant cells. The Cag-T4SS is adapted for pathogenicity in humans and plays a critical role in gastric carcinogenesis. Despite its significance, the molecular details governing the assembly and function of the Cag-T4SS, particularly the inner membrane core complex (IMCC), remain partly characterized.
In this study, we identified and characterized a direct and specific interaction between CagF and CagV, two essential components of the Cag-T4SS. Using a combination of in silico molecular docking, MD simulations, and in vitro assays, including a GST pull-down experiment, we mapped the key binding region of CagF to residues 51-100 at its N-terminus. These findings provide the first molecular insight into the CagF-CagV interface and suggest a functional role for this interaction in the stepwise assembly of the IMCC. Our results extend previous work showing that CagV, a bitopic inner membrane protein, connects the IMCC and OMCC through interactions with CagX and CagZ.16 We propose that the newly characterized CagF-CagV interaction implies that CagV may act as an anchoring scaffold for the CagF-CagA complex, positioning it optimally near the secretion channel. This spatial organization is likely critical for the efficient recruitment of CagA to the secretion conduit, coordinated by ATPases such as CagE, Caga, and Cagb. A proposed sequential assembly model of the Cag-ATPases within the IMCC suggests that the VirB4-like ATPase CagE is initially recruited to the IMCC, establishing the foundation for subsequent ATPase incorporation.9 This is followed by docking of the VirB11-like ATPase Caga, and finally the recruitment of the VirD4-like coupling ATPase Cagb, which collectively form the central energy-transducing hub essential for substrate translocation. Notably, CagZ binds directly to Cagb and acts as a negative regulator of its ATPase activity.47,48 Structural studies have shown that CagZ binding prevents Caga from forming its functional hexameric conformation, which is necessary for ATP hydrolysis and energy supply to the secretion machinery. Release of CagZ appears to be a critical step that enables Cagb to oligomerize and activate, thereby completing the ATPase module required for secretion. This proposed assembly sequence is supported by structural analyses, including studies employing deletion mutants of the individual ATPases. It has been reported that CagF functions as a dedicated chaperone14 that maintains CagA in a secretion-competent state by binding near its C-terminal translocation signal.15 The subsequent dissociation of CagF from CagA may facilitate the exposure of the translocation signal, enabling direct interaction with Cagb and promoting CagA’s passage through the secretion channel. Interestingly, prior immunoprecipitation studies found inconsistent enrichment of IMCC components when hemagglutinin-tagged CagF (HA-CagF) was used as bait, suggesting that the CagF-CagA complex may engage only transiently with the secretion machinery or that additional regulatory factors influence these interactions.49 Furthermore, the relatively large size of CagF compared to classical secretion chaperones raises the possibility of broader regulatory roles beyond merely stabilizing CagA. Although our data suggest that the CagF-CagV interaction does not directly stabilize CagF (as CagF expression was unaffected by the absence of CagV), this complex likely contributes to efficient effector delivery by organizing CagA close to the energy-providing ATPase complex.
From a therapeutic perspective, mapping the interacting residues within the CagF-CagV complex creates opportunities to disrupt this interaction selectively. MD analysis and future machine learning-based approaches50 could explore the binding of active compounds, such as quercetin, kaempferol, and luteolin, to these critical residues. Additionally, natural compounds derived from Terminalia chebula Retz, such as chebulinic acid and 1,3,6-trigalloylglucose, have shown potential to suppress CagA expression and interfere with H. pylori adhesion,51,52 representing promising adjunct strategies against antibiotic-resistant strains. Together, our findings advance the understanding of the CagF-CagV interaction as an integral molecular event in the assembly and function of the Cag-T4SS.
This study provides compelling evidence for a physical interaction between CagF and CagV, which likely plays a pivotal role in facilitating the translocation of the effector protein CagA through the Cag-T4SS. Co-localization studies revealed that CagF and CagV form distinct foci at the inner membrane of H. pylori, suggesting spatial proximity that reinforces their physical interaction. Computational analysis, combined with biochemical assays, further validated this interaction, identifying the N-terminal region of CagF as the primary binding interface. Notably, residues 51-100 at the N-terminus of CagF were identified as critical for CagF-CagV interaction, highlighting their potential significance in the structural integrity and functional assembly of the IMCC. This interaction appears to ensure the spatial and temporal recruitment of CagA to the secretion channel and facilitate ATPase-dependent translocation, critical steps in H. pylori induced pathogenesis. Further targeted mutagenesis and high-resolution structural studies will be invaluable to fully elucidate the molecular mechanisms and therapeutic potential of this interaction.
Additional file: Additional Figure S1-S3.
ACKNOWLEDGMENTS
The authors extend their sincere gratitude to Prof. Gauranga Mukhopadhyay, Jawaharlal Nehru University, New Delhi, India, for his valuable scientific input and for providing the H. pylori strains and primary antibodies used in this study, and to Dr. Arjun Ray and Dr. Abhishek Mukherjee for their support. The authors also acknowledge Jawaharlal Nehru University, New Delhi, Gautam Buddha University, Greater Noida, and Institute of Information Technology-Delhi, New Delhi, for their infrastructural facilities and technical support.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
AUTHORS’ CONTRIBUTION
NK conceptualized the study and collected resources. SSai applied methodology. SSai, SSac and NK performed investigation. SSai, SSac, BS and NK performed formal analysis. SSai and NK performed data validation and data curation. SSai, SSac and NK performed visualization. NK performed supervision and project administration. SSai, SSac and NK wrote the original draft. SSai, BS and NK wrote, reviewed and edited the manuscript. All authors read and approved the final manuscript for publication.
FUNDING
None.
DATA AVAILABILITY
The datasets generated and/or analysed during the current study are available from the corresponding author on reasonable request.
ETHICS STATEMENT
Not applicable.
- World Health Organisation (WHO). Schistosomes, liver flukes and Helicobacter pylori. IARC Monogr Eval Carcinog Risks Hum. 1994;61:1-241.
- Fischer W, Puls J, Buhrdorf R, Gebert B, Odenbreit S, Haas R. Systematic mutagenesis of the Helicobacter pylori cag pathogenicity island: Essential genes for CagA translocation in host cells and induction of interleukin-8. Mol Microbiol. 2001;42(5):1337-1348.
Crossref - Sain S, Solanki B, Kumar N. Helicobacter pylori CagA and CagT antibodies arrest the translocation of CagA into gastric epithelial cells. 3 Biotech. 2025;15(6):179.
Crossref - Ou L, Liu H, Peng C, et al. Helicobacter pylori infection facilitates cell migration and potentially impact clinical outcomes in gastric cancer. Heliyon. 2024;10(17):e37046.
Crossref - Kumar N, Sain S, Solanki B. Deciphering Microbial Carbohydrate Metabolism: Unveiling Mechanistic Insights Into Type 2 Diabetes. Adv Gut Microb Res. 2025;2025(1):4395850.
Crossref - Duan Y, Xu Y, Dou Y, Xu D. Helicobacter pylori and gastric cancer: mechanisms and new perspectives. J Hematol Oncol. 2025;18(1):10.
Crossref - Fazio A, Bitran-Ambler M, Ramirez-Rivera S, Zaffiri V, Bernal G. Genotyping of Helicobacter pylori CagA/CagE strains in gastric mucosa and its association with gastric illness. Diagn Microbiol Infect Dis. 2023;107(2):116028.
Crossref - Gou L, Yang X, Yun J, et al. Roles of the components of the cag-pathogenicity island encoded type IV secretion system in Helicobacter pylori. Future Microbiol. 2024:1-15.
Crossref - Hu B, Khara P, Song L, et al. In Situ Molecular Architecture of the Helicobacter pylori Cag Type IV Secretion System. mBio. 2019;10(3).
Crossref - Frick-Cheng AE, Pyburn TM, Voss BJ, McDonald WH, Ohi MD, Cover TL. Molecular and Structural Analysis of the Helicobacter pylori cag Type IV Secretion System Core Complex. mBio. 2016;7(1):e02001-15.
Crossref - Kumari R, Shariq M, Kumar N, Mukhopadhyay G. Biochemical characterization of the Helicobacter pylori Cag type IV secretion system unique component CagU. FEBS Lett. 2017;591(3):500-512.
Crossref - Shariq M, Kumar N, Kumari R, Kumar A, Subbarao N, Mukhopadhyay G. Biochemical Analysis of CagE: A VirB4 Homologue of Helicobacter pylori Cag-T4SS. PLoS One. 2015;10(11):e0142606.
Crossref - Kumari R, Shariq M, Sharma S, Kumar A, Mukhopadhyay G. CagW, a VirB6 homologue interacts with Cag-type IV secretion system substrate CagA in Helicobacter pylori. Biochem Biophys Res Commun. 2019;515(4):712-718.
Crossref - Couturier MR, Tasca E, Montecucco C, Stein M. Interaction with CagF Is Required for Translocation of CagA into the Host via the Helicobacter pylori Type IV Secretion System. Infect Immun. 2006;74(1):273-281.
Crossref - Pattis I, Weiss E, Laugks R, Haas R, Fischer W. The Helicobacter pylori CagF protein is a type IV secretion chaperone-like molecule that binds close to the C-terminal secretion signal of the CagA effector protein. Microbiology (NY). 2007;153(9):2896-2909.
Crossref - Kumar N, Shariq M, Kumar A, et al. Analyzing the role of CagV, a VirB8 homolog of the type IV secretion system of Helicobacter pylori. FEBS Open Bio. 2017;7(7):915-933.
Crossref - Kumar N, Shariq M, Kumari R, Tyagi RK, Mukhopadhyay G. Cag Type IV Secretion System: CagI Independent Bacterial Surface Localization of CagA. PLoS One. 2013;8(9):e74620.
Crossref - Tolia NH, Joshua-Tor L. Strategies for protein coexpression in Escherichia coli. Nat Methods. 2006;3(1):55-64.
Crossref - LAEMMLI UK. Cleavage of Structural Proteins during the Assembly of the Head of Bacteriophage T4. Nature. 1970;227(5259):680-685.
Crossref - Cendron L, Couturier M, Angelini A, Barison N, Stein M, Zanotti G. The Helicobacter pylori CagD (HP0545, Cag24) Protein Is Essential for CagA Translocation and Maximal Induction of Interleukin-8 Secretion. J Mol Biol. 2009;386(1):204-217.
Crossref - Hallgren J, Tsirigos KD, Pedersen MD, et al. DeepTMHMM predicts alpha and beta transmembrane proteins using deep neural networks. 2022.
Crossref - Wu X, Zhao Y, Sun L, et al. Crystal structure of CagV, the Helicobacter pylori homologue of the T4SS protein VirB8. FEBS J. 2019;286(21):4294-4309.
Crossref - Honorato RV, Trellet ME, Jimenez-Garcia B, et al. The HADDOCK2.4 web server for integrative modeling of biomolecular complexes. Nat Protoc. 2024;19(11):3219-3241.
Crossref - Honorato RV, Koukos PI, Jimenez-Garcia B, et al. Structural Biology in the Clouds: The WeNMR-EOSC Ecosystem. Front Mol Biosci. 2021;8:729513.
Crossref - Hubbard SJ, Thornton JM. ‘NACCESS’, computer program. Department of Biochemistry and Molecular Biology, University College, London. 1993.
- Rodrigues JPGLM, Trellet M, Schmitz C, et al. Clustering biomolecular complexes by residue contacts similarity. Proteins. 2012;80(7):1810-1817.
Crossref - Vangone A, Rodrigues JPGLM, Xue LC, et al. Sense and Simplicity in HADDOCK Scoring: Lessons from CASP CAPRI (page 418). [published correction appears in Proteins. 2017;85(8) 1589-1590] Proteins. 2017;85(3): 417-423.
Crossref - Sondergaard CR, Olsson MHM, Rostkowski M, Jensen JH. Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pKa Values. J Chem Theory Comput. 2011;7(7):2284-2295.
Crossref - Olsson MHM, Sondergaard CR, Rostkowski M, Jensen JH. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J Chem Theory Comput. 2011;7(2):525-537.
Crossref - Jurrus E, Engel D, Star K, et al. Improvements to the APBS biomolecular solvation software suite. Protein Science. 2018;27(1):112-128.
Crossref - Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera-A visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605-1612.
Crossref - Laskowski RA, Jablonska J, Pravda L, Varekova RS, Thornton JM. PDBsum: Structural summaries of PDB entries. Protein Sci. 2018;27(1):129-134.
Crossref - Bekker H, Berendsen HJC, Dijkstra EJ, et al. Gromacs – A Parallel Computer for Molecular-Dynamics Simulations. In: Degroot RA, Nadrchal J, (eds). Physics Computing ’92. World Scientific Publishing; 1993:252-256.
- Berendsen HJC, van der Spoel D, van Drunen R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput Phys Commun. 1995;91(1-3):43-56.
Crossref - Lindahl E, Hess B, van der Spoel D. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J Mol Model. 2001;7(8):306-317.
Crossref - Best RB, Zhu X, Shim J, et al. Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone φ, ψ and Side-Chain c1 and c2 Dihedral Angles. J Chem Theory Comput. 2012;8(9):3257-3273.
Crossref - Klauda JB, Venable RM, Freites JA, et al. Update of the CHARMM All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. J Phys Chem B. 2010;114(23):7830-7843.
Crossref - Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79(2):926-935.
Crossref - Hess B, Bekker H, Berendsen HJC, Fraaije JGEM. LINCS: A linear constraint solver for molecular simulations. J Comput Chem. 1997;18(12):1463-1472.
Crossref - Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. Molecular dynamics with coupling to an external bath. J Chem Phys. 1984;81(8):3684-3690.
Crossref - Valdes-Tresanco MS, Valdes-Tresanco ME, Valiente PA, Moreno E. gmx_MMPBSA: A New Tool to Perform End-State Free Energy Calculations with GROMACS. J Chem Theory Comput. 2021;17(10):6281-6291.
Crossref - Miller BR, McGee TD, Swails JM, Homeyer N, Gohlke H, Roitberg AE. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J Chem Theory Comput. 2012;8(9):3314-3321.
Crossref - Waterhouse A, Bertoni M, Bienert S, et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46(W1):W296-W303.
Crossref - Rosilan NF, Jamali MAM, Sufira SA, et al. Molecular docking and dynamics simulation studies uncover the host-pathogen protein-protein interactions in Penaeus vannamei and Vibrio parahaemolyticus. PLoS One. 2024;19(1):e0297759.
Crossref - Gasteiger EBA, Hoogland C, Gattiker. A, et al. Protein Identification and Analysis Tools on the ExPASy Server. In: Walker JM. (eds) The Proteomics Protocols Handbook. Springer Protocols Handbooks. Humana Press. 2005.
Crossref - Jiang L, Lai L. CH···O Hydrogen Bonds at Protein-Protein Interfaces. Journal of Biological Chemistry. 2002;277(40):37732-37740.
Crossref - Wu X, Zhao Y, Zhang H, et al. Mechanism of regulation of the Helicobacter pylori Cagb ATPase by CagZ. Nat Commun. 2023;14(1):479.
Crossref - Jurik A, Hauber E, Kutter S, et al. The Coupling Protein Cagb and Its Interaction Partner CagZ Are Required for Type IV Secretion of the Helicobacter pylori CagA Protein. Infect Immun. 2010;78(12):5244-5251.
Crossref - Bryant KN, Frick-Cheng AE, Solecki LE, et al. Species-specific components of the Helicobacter pylori Cag type IV secretion system. Infect Immun. 2025;93(5):e0049324.
Crossref - Xu Z, Rasteh AM, Dong A, Wang P, Liu H. Identification of molecular targets of Hypericum perforatum in blood for major depressive disorder: a machine-learning pharmacological study. Chin Med. 2024;19(1):141.
Crossref - Ou L, Zhu Z, Hao Y, et al. 1,3,6-Trigalloylglucose: A Novel Potent Anti-Helicobacter pylori Adhesion Agent Derived from Aqueous Extracts of Terminalia chebula Retz. Molecules. 2024;29(5):1161.
Crossref - Ou L, Hao Y, Liu H, et al. Chebulinic acid isolated from aqueous extracts of Terminalia chebula Retz inhibits Helicobacter pylori infection by potential binding to Cag A protein and regulating adhesion. Front Microbiol. 2024;15.
Crossref
© The Author(s) 2025. Open Access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License which permits unrestricted use, sharing, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.