


ISSN: 0973-7510
E-ISSN: 2581-690X
Our study aimed at metagenomic exploration of effluent contaminated soils across Dadra and Nagar Haveli region, India. Physico-chemical analysis of soil samples SA4, SA5, and SA6 revealed that the presence of macro and micro elements which includes metals and non-metals. Furthermore, it includes various petroleum and other aromatic hydrocarbons in it. Metagenomic sequencing was followed by taxonomic assessment and functional annotation. Taxonomic assessment demonstrated dominance of Pseudomonas and Bacillus species in all SA4, SA5, and SA6 samples. Moreover, it includes diverse microbial communities that are involved in degradation of xenobiotic compounds which includes Sulfuricurvum kujiense, Novosphingobium sp., Variovorax paradoxus, Usitatibacter rugosus, Cupriavidus sp. and many others found in soil samples. Functional annotation like KEGG and COG, and found a range of hydrocarbon degrading enzymes that are involved with benzoate derivatives. A comprehensive metabolic network that outlines the degradation pathways for aromatic hydrocarbons-like benzoate, chlorobenzoate, polycyclic aromatic hydrocarbons, xylene, toluene, ethylbenzene, nitrotoluene, aminobenzoate, chloroalkane, and chloroalkene and the derivative of these compounds were furtherly entered into TCA cycle. Presence of such microbial species and their genes will be helpful for further development of bioremediation strategies to remediate such contaminated sites.
Bioremediation, COG, KEGG, Metagenomics, Xenobiotics
Nowadays, significant growth in industrialization has contributed to sustainable economic growth and advancement in novel technologies but has also led to significant environmental degradation, which resulted into wide ranges of environmental effects, especially in soil contamination. Industrial effluents, consist of mixture of hazardous compounds such as toxic elements, chemicals, metals and various organic compounds, which directly affects the soil ecosystems.1,2 Xenobiotics, which often consist of aromatic and aliphatic hydrocarbons are the major compounds amongst these, which are seems to having major concern as their potential persistence in the terrestrial as well as in marine ecosystem.3 These hydrocarbons are extensively used in various industrial process such as plastic manufacturing, petroleum extraction, pharmaceuticals, surfactant industries and discharge of these effluent in soil system may leads to various ecological and health risks which may resulted into deterioration of terrestrial and microbial flora of the soil quality which are involve in significant geological cycles.4,5 For development of novel strategies for bioremediation of such pollutants, detailed understanding of microbial communities present in such contaminated soil is required.4 As, these microbial communities have significant versatility to metabolically transform and degrade such pollutants.6
Soil microbiota plays essential role in various geobiological cycles and in maintaining ecosystem functionality. Diverse microbial communities can enhance the resistance to number of environmental factors, facilitate nutrient availability, and promote plant growth.7 However, discharge of such industrial effluents into soil can leads to significant alteration in microbial diversity as well as their functionality.8 The loss of microbial diversity may result into significant adverse effects on soil health, reduced productivity and increased susceptibility to pathogens.
To study microbial communities in In vitro using traditional cultured based methods, it is limited up to certain level as microbial communities are diverse in nature and all are not cultivable in laboratory.9 Consequently, these methods can overlook numerous unculturable or rare taxa, restricting our understanding of functional diversity. The emergence of metagenomic sequencing has unveiled potential alternative which allows in depth analysis of microbial communities by isolating nucleic acid and sequencing of genetic material directly from the environmental samples, researchers can gain insights into the entire microbial community structure, their functional potential, and interactions among diverse microbial taxa.10
High-throughput metagenomic studies reveals that diverse microbial communities exhibits in such contaminated soils having unique compositional characteristic and gene profiles related to pollutants degradation. Previous studies indicates that certain microbial taxa are adapted in specific industrial contaminants and having different pathways for degradation of xenobiotic compounds11 and as such, corelating the relationship amongst these bacterial communities and their functional abilities to degradation of aromatic and aliphatic hydrocarbons is significantly helpful for devising effective bioremediation strategies.
Aromatic and aliphatic compounds are the major components of many industrial wastes, which includes phenols, benzene, toluene, ethylbenzene and xylene.12 As such, these compounds are not only toxic but they are hydrophobic in nature which directly makes difficult for bioremediation. As they are discharged in natural environment, they can remain present for long term period, and directly or indirectly affecting soil microbial flora, as well as plants and animals, respectively.13,14
Recent studies indicates that these xenobiotic compounds may undergo bioaccumulation in the food chain, leading to long term ecological consequences and exposure for human populations.15 Given these critical issues, naturally microorganism is capable to degrade these compounds in contaminated soil reduces their impact in environment and restoring the ecosystem health.
Microorganism uses different pathways for metabolism of xenobiotic compounds like aromatic and aliphatic hydrocarbons which includes cumulative pathways under aerobic and anaerobic conditions, hydrolysis, oxidation and other biochemical transformations.6,16 Different metabolic pathways have been identified, illustrating the microbial communities were capable to degrade diverse xenobiotic compounds such as ortho and meta-cleavage pathways for degradation of xenobiotic compounds used by different microbial genus like Pseudomonas, Rhodococcus and Sphingomonas, respectively.17 Similarly, nβ-oxidation pathways are used by specialized group of microorganisms for aliphatic hydrocarbon degradation into simpler non-toxic components.3 Furthermore, the synergistic interactions among microbial taxa can enhance degradation rates, as different species may possess complementary metabolic capabilities.18 For example, one species may transform a xenobiotic compound into a more biodegradable form, while another may utilize that compound as a substrate for growth. Understanding these interactions at the genomic level through metagenomic exploration is essential for unraveling the complex dynamics of microbial communities involved in degradation of such xenobiotics.
The advancement of metagenomic sequencing combines with metabolomics and meta proteomics has recently offered an alternative for in-depth investigation of microbial community and their metabolic potential.19,20 Many studies have employed large-scale metagenomic sequencing to examine the human gut microbiota,21 and marine microbiome.22 A lots of research attention was also paid to the high throughput metagenomic analysis of soils because of their complicated features, which are controlled by a variety of significant variables.23 The recent advancement in bioinformatics also helpful insight depth investigation to unveiled microbial communities and their genes involve in various aromatic hydrocarbon degradation.24
The aim of the study is to evaluate genomic and native microbial communities’ capabilities to possess different genes and enzymes required for the degradation and metabolism of different xenobiotic compounds present in contaminated soil samples. We seek to explore, from a genome-wide perspective, genes and enzymes of microbial communities and metabolic pathways of the bacteria residing in industrial effluent contaminated soils. This investigation will enhance our understanding of the soil metagenome in terms of its genetic content and functional attributes.
Collection of industrial effluent contaminated soil samples
Soil sampling was performed systematically to ensure accurate representation of industrial effluent contaminated areas of Dadra and Nagar Haveli, India. The sampling methodology followed standard protocols to minimize bias and ensure reliability. Sampling locations were determined based on historical data and field surveys. Samples were collected on 28th of August 2023 from three different locations. SA4 collected from: Hindustan Unilever Ltd, Dapada, D&NH, Silvassa, (20.182935676187814°N and 73.03031053814586°E). SA5 samples were collected from Arti Industrie Pvt. Ltd., Kherdi, D & NH, Silvassa (20.097537416773758°N and 73.01687295944654°E). SA6 sample were collected from Hind Aluminum Industries Pvt. Ltd., Dapada, D&NH, Silvassa (20.186875541268474°N and 73.02571179752846°E) at depths of 5-15 cm to evaluate the vertical distribution of contaminants, as recommended by the Bao et al.25 At each site, approximately 500 grams of soil was collected using a clean stainless-steel trowel to prevent cross-contamination. The soil was placed into pre-labeled SA4, SA5, and SA6, sterile polyethylene bags. Multiple subsamples were taken from different points within a 1-meter radius to account for spatial variability.26 All Soil samples were kept in cold chamber at 4 °C during transport research laboratory to minimize changes in soil chemistry. Analysis was performed within 48 hours of collection to maintain sample integrity.27
Soils physiochemical analysis
Soil samples weighing approximately 500 g were collected from each of the industrial effluent contaminated site by randomly selecting 2 to 3 locations in proximity to the rhizosphere zone. The samples from these locations were combined, and a representative 500 g sample was extracted as previously outline (https://cals.arizona.edu/back yards/sites/cals.arizona.edu.backyards/files/p16-17). Of the collected samples, approximately 150 g was allocated for soil metagenomic analysis, while the remaining 350 g was designated for assessing soil physiochemical properties. Each sample underwent analysis according to established protocols for various physio-chemical characteristics including pH, conductivity, nitrogen (N2), organic carbon, organic matter, total soluble solids, COD, and BOD (IS), available sulphur, chloride (Cl), moisture content, water holding capacity and phosphorous (P)(FL/SOP/S-05), boron (B), calcium (Ca), cobalt (Co), copper (Cu), magnesium (Mg), manganese (Mn), molybdenum(Mb), potassium (K), iron (Fe), zinc (Zn) (FL/SOP/12-25) and petroleum aromatic hydrocarbons were detected using GC-MS (FL/SOP/02-104) (S.K Maiti, Handbook of Methods in Environmental Studies Vol.1) and (Method for sampling and analysis of dissolved methane in wastewater by G.Tjandraatmadja, D.Beale, N.Goodman, M.Toifl and D.Marney).28
DNA extraction and quantification
The metagenomic nucleic acid was extracted from all three SA4, SA5 and SA6 soil samples by using PowerSoil DNA Isolation Kit (MO BIO’s Ultraclean® Soil DNA Laboratories Inc., USA) as per instruction given in manual. Approximately 0.25 g of soil was weighed and added into bead beating tube containing 60 µL of C1 solution and vortex for 10 min at maximum speed to lyse microbial cells, followed by transferring supernatant into 2 ml collection tube. The subsequent steps involved centrifugation, binding of DNA to the provided spin filter, washing with the wash buffer, and finally elution of DNA is carried by adding 25 µL of elution buffer (MO BIO’s PowerSoil DNA Isolation Kit Ultraclean® Soil DNA Laboratories Inc., USA). Quantification of the extracted DNA using a nanodrop spectrophotometer (Thermo Scientific, Wilmington, DE, USA). The DNA concentration was determined at 260 nm, while purity was evaluated by calculating the absorbance ratio at 260 nm to 280 nm (A260/A280) (Table S1). Samples with a ratio between 1.8 and 2.0 were considered pure.29 Additionally, we assessed DNA integrity using agarose gel electrophoresis. We loaded 5 µL of each sample onto a 1% agarose gel that was stained with ethidium bromide and visualized it under UV light, as illustrated in (Molecular cloning: a laboratory manual/Michael R. Green, Joseph Sambrook.-4th ed) (Figure S1).30
Construction of DNA library and performing metagenomic sequencing
Construction of DNA library
The whole nucleic acid concentration and quality of DNA was analyzed by radially available sophisticated instrument (Thermo Fisher Scientific, Waltham, MA, USA), i.e. Qubit fluorometer and a Nanodrop spectrophotometer. DNA library was prepared using Nextera XT DNA Library Preparation Kit (Illumina, San Diego, CA, USA). For that 1 ng of DNA was used for the library construction. The protocol included fragmentation, amplification via PCR, and purification of the library using magnetic beads. 4200 Tape station system (Agilent Technologies) were used to analyzed PCR enriched DNA libraries as per instruction manual provided by manufacturer using high sensitivity D1000 Screen tape system.
Sequencing
DNA libraries were quantified using nanodrop spectrophotometer and diluted to a concentration of 10 nmol prior to sequencing. Metagenomic sequencing was executed utilizing the Illumina MiSeq Platform, employing a 2 x 250 bp paired-end read configuration. The WGS process was carried out in accordance with the manufacturers protocols, resulting in the generation of raw data in FASTAQ format (Illumina, San Diego, CA, USA, 2020).31
Raw data processing and Bioinformatic analysis
Raw sequencing reads was analyzed by using software package QIIME 2.32 This included quality filtering using MEGAHIT,33 trimming using Trimmomatic v0.39,34 and operational taxonomic unit (OTU) clustering at 97% similarity a (MaxBin2 binner). Silva reference database was used to assess the taxonomic profiling.35 Followed by R software was used to analyzed statistical and microbial diversity composition of microbial communities.36
Taxonomic assessment
Taxonomic profiling and classification were conducted on the assembled genome utilizing37 Kraken2 v2.0.9. This tool is recognized for its high classification accuracy and computational efficiency, rendering it particularly effective for extensive metagenomic analyses. It incorporates a range of methodologies, including optimized data structures and parallel processing, to handle sequencing data with efficiency. The tool relies on a standard RefSeq database that encompasses taxonomic information pertaining to archaea, bacteria, viruses, plasmids, humans, fungi, and vector-associated species.
Functional annotation
Functional annotation of the genes of SA4, SA5, and SA6 was conducted utilizing eggnog, a robust stand-alone framework capable of concurrently providing COG, KEGG, and Pfams subsystem annotation for individual sequences within metagenomic databases. The incorporation of a novel ‘directed search’ mechanism in eggnog significantly diminishes the computational demands typically associated with functional analysis.38 KEGG, COG, orthologous groups (KOs) analysis were used to categorize proteins into different orthologous groups and furtherly it was used to construct different metabolic pathways using KEGG analysis.39,40
Sequence deposition and data accession
The whole metagenomic sequence data of these 3 samples were submitted in NCBI databases under the NCBI Bioproject number PRJNA1084772. The metagenomic sequencing data is available in NCBI Sequence Read Archive under accession numbers SAMN46715193 (SA4 soil sample), SAMN46715194 (SA5 soil sample), SAMN46715195 (SA6 soil sample).
Physiochemical analysis of industrial effluent contaminated soil samples
Soil samples SA4, SA5, and SA6 exhibited notable variations in edaphic properties, including moisture level, pH, total phosphorus content, conductivity, organic matter, available sulfur, chloride, and carbon-to-nitrogen (C:N) ratio (Table S2). The soils samples from industrial effluent contaminated sites were characterized by having slightly neutral to basic pH, the second highest high conductivity, high phosphorous, followed by high water holding capacity, moisture content and the lowest of organic matter, carbon, and nitrogen, available chloride, total soluble solids and available sulfur content. Similarly, the micro and macronutrients including metals and heavy metal concentration in collected soil samples SA4, SA5, and SA6 from different location are presented in Table S2. The results of pairwise comparisons differed based on the specific type of minerals and metals; nonetheless, it was observed that metals such as iron (Fe) (95.69 mg/Kg), (114.11 mg/Kg), and (121.31 mg/Kg), followed by calcium (Ca) (51.42 mg/Kg), (70.35 mg/Kg), and (72.31 mg/Kg), magnesium (Mg) having (14.17 mg/Kg), (15.77 mg/Kg), and (19.16 mg/Kg), potassium (K) having (10.18 mg/Kg), (15.13 mg/Kg), and (14.45 mg/Kg) in SA4, SA5, and SA6, respectively. Some other elements, like manganese (Mn), copper (Cu), and zinc (Zn) are also available in trace amount (Table S2). Further, each soil sample was analyzed for petroleum and other aromatic hydrocarbons with GC-MS which indicates that various aromatic hydrocarbons like acetone, acetonitrile, ethanolamine, 2-ethoxyethanol, and other are present in respective samples (Table S2). SA4, SA5, and SA6 exhibits higher concentration of macronutrients, including carbon (C), phosphorus (P), which significantly affected the microbial community’s structure. The organic carbon levels were measured at 2.60%, 1.39%, and 1.33% in SA4, SA5, and SA6, respectively. Our finding suggest that the increased carbon levels in these specimens are caused by the existence of aromatic and aliphatic organic hydrocarbons in polluted soil. Elevated carbon levels in these samples are attributed to the presence of aromatic organic hydrocarbons in the contaminated soil. Industrial effluent and discharge of wastewater in soil are the major source of these pollutants like polycyclic aromatic hydrocarbons (PAHs) and polychlorinated biphenyls (PCBs). Several studies confirms that soils exhibiting high microbial diversity tends to have elevated phosphorus (P) and low carbon to phosphorus (C/P) ratios.41-44 The availability of inorganic nutrients plays significant role in both structural and catalytic functions. Consequently, the overall microbial community structure is largely influenced by the presence/absence of carbon, nitrogen, along with the availability of inorganic nutrients like calcium, potassium, magnesium, phosphorus to some degree.45 Metal analysis indicates the presence of metals like Zn, Cu, Fe in higher concentration while trace amount of Mn creates an appropriate environment for microbes to engage in anaerobic degradation. The existence of these micro and macro nutrients soil samples can hinder the growth of various microbial species, while simultaneously promoting the growth of microbial species capable to tolerate and metabolize such metals, if they are present in higher concentration as well. The findings aligns with numerous studies that emphasize that the accumulation of these heavy metals and toxic xenobiotic pollutants like polycyclic aromatic hydrocarbons and PCB, in soils associated with iron and steel, petrochemical industries.44,46
Whole Metagenomic Sequencing and Gene Identification
Whole metagenomic sequencing was performed using the Illumina MiSeq Platform, resulting in the acquisition of 8.9 million, 10.6 million and 13.9 million sequencing reads of SA4, SA5, and SA6 and producing 2.43 Gb, 3.2 Gb and 4.16 Gb of nucleotide data, respectively. De novo assembly of nucleotide sequence was done to obtained gene orthologs, using MEGAHIT [MetaWRAP pipeline v1.3.2]47 (Table S3). Further, the gene were predicted from the binned scaffolds of all samples using Metagene Mark48 with default parameters (Table S4). The presence of genes encoding for various hydrocarbon degradation like catE, dmpB, xylE, pcaH; pcaG, ligA, ligB, ligI, tmoE, tbuA2, touE, nemA, etbD, chnB, tpmT, kstD, catA, catB, pcaC, bcrB, badE, hcrC, hbaB, hbaC, hmfF, pht3, dehH, hyaB, hybC, hsaC and vgb which are responsible for degradation of benzoate, aminobenzoate, flurobenzoate, toluene, nitrotoluene, chlorocychlohexane and chlorobenzene, caprolactam, ethylebenzene, polycyclic aromatic hydrocarbon, furfural and xylene were represented in Table. The identification of genes responsible for anaerobic degradation through benzoyl-CoA (badA, hbaA, hbaB, hcrC) as well as those for aerobic degradation via protocatechuate (ligA, ligC, ligI, pobA, pcaB), catechol (catE) indicates a comprehensive pathway for benzoate degradation.44 Additionally, some studies reported that some of facultative anaerobes like denitrifying Thauera, Magnetospirillum strains, and the photoheterotroph Rhodospeudomonas were capable to degrade benzoate via benzoyl-CoA pathway under anaerobic conditions.44,49 This reported gene supports the earlier finding of Weiland-Brauer et al. and Isaac et al.43,50
Table:
Genes and enzymes with microbial species responsible for degradation of xenobiotic compounds of SA4, SA5, and SA6
| No. | Genes and Enzymes | Microbial species | Numbers of sequence present in each metagenome | ||
|---|---|---|---|---|---|
| SA4 | SA5 | SA6 | |||
| 1 | E3.1.1.45; Carboxymethylenebutenolidase(2) | Pseudomonas aeruginosa, Cupriavidus necator, | 2 | 7 | 9 |
| 2 | catE, dmpB, xylE; Catechol 2,3-dioxygenase(7) | Pseudomonas putida, Sphingomonas paucimobilis, Acinetobacter sp., Alcaligenes sp. strain O-1, Geobacter metallireducens, Rhodopseudomonas palustris, Syntrophus aciditrophicus, Cupriavidus necator | 8 | 11 | 24 |
| 3 | bla2, blm, ccrA, blaB; Metallo-beta-lactamase class B | Pseudomonas aeruginosa, Neisseria meningitidis PorB., Gonococcal, Bacillus licheniformis, Acinetobacter baumannii, Staphylococcus aureus, Enterococcus faecium | 1 | 6 | |
| 4 | E1.14.13.40; anthraniloyl-CoA monooxygenase (2) | Pseudomonas putida ATCC 12633., Pseudomonas putida strain A10L, Rhodococcus rhodochrous, Cupriavidus necator | 2 | 5 | 8 |
| 5 | Aminoglycoside 6-adenylyltransferase | Serratia marcescens, uncultured bacterium, Campylobacter jejuni, Cupriavidus necator | 1 | 1 | |
| 6 | pcaH; pcaG, ligA, ligB, ligI; Protocatechuate 3,4-dioxygenase (7) | Sphingomonas paucimobilis SYK-6, Alcaligenes sp. strain O-1, Geobacter metallireducens, Acinetobacter sp. strain ADP1, Rhodopseudomonas palustris, Cupriavidus necator | 7 | 9 | |
| 7 | tmoE, tbuA2, touE; Toluene monooxygenase (3) | Pseudomonas stutzeri OX1, Thauera aromatica, Denitrifying bacterium strain EbN1, Comamonas testosteroni, Alcaligenes sp. strain O-1. | 3 | 1 | |
| 8 | E3.8.1.2; 2-haloacid dehalogenase (4) | Pseudomonas sp. strain YL, Moraxella sp. strain B | 4 | ||
| 9 | nemA; N-ethylmaleimide reductase (8) | Pseudomonas pseudoalcaligenes JS52., Pseudomonas aeruginosa Strain., Burkholderia sp. strain DNT. | 8 | ||
| 10 | etbD; 2-hydroxy-6-oxo-octa-2,4-dienoate hydrolase(2) | Rhodococcus sp. strain RHA1. | 2 | ||
| 11 | chnB; Cyclohexanone monooxygenase (2) | Acinetobacter NCIB 9871., Xanthobacter sp., Pseudomonas species. | 2 | 11 | |
| 12 | tpmT; Thiopurine S-methyltransferase(2) | Mycobacterium sp. | 2 | ||
| 13 | kstD; 3-oxosteroid 1-dehydrogenase(2) | Mycobacterium tuberculosis CYP142, Comamonas testosterone | 2 | ||
| 14 | catA, catB; Chloramphenicol O-acetyltransferase (11) | Pseudomonas aeruginosa, Neisseria meningitidis PorB., Gonococcal, Bacillus licheniformis., Acinetobacter baumannii | 11 | ||
| 15 | pcaC; 4-carboxymuconolactone decarboxylase (3) | Pseudomonas stutzeri OX1, Thauera aromatica, Denitrifying bacterium, strain EbN1, Comamonas testosteroni, Alcaligenes sp. strain O-1. | 3 | ||
| 16 | bcrB, badE; Benzoyl-CoA reductase(6) | Clostridium botulinum, Thauera aromatica and Cryptobacterium curtum | 6 | ||
| 17 | hcrC, hbaB, hcrA, hbaC; 4-hydroxybenzoyl-CoA reductase (4) | Pseudomonas species, Thauera aromatica, | 4 | ||
| 18 | hmfF; 2,5-furandicarboxylate decarboxylase (1) | Cupriavidus basilensis HMF14., | 1 | ||
| 19 | cmtAc; p-cumate 2,3-dioxygenase(1) | Rhodococcus sp. strain 19070, Beijerinckia sp. strain B1, Desulfotomaculum, Pseudomonas putida F1 | 1 | ||
| 20 | pht3; phthalate 4,5-dioxygenase | Pseudomonas putida strains G7, Burkholderia sp., Comamonas sp. strain JS765., Mycobacterium sp., Mycobacterium vanbaalenii PYR-1. | 2 | ||
| 21 | dehH; Haloacetate dehalogenase (1) | Dehalococcoides sp., Xanthobacter autotrophicus GJ10, Pseudomonas sp. strain DCA1, Pseudomonas cichorii 170, Mycobacterium sp. TA27. | 1 | ||
| 22 | hyaB, hybC; Hydrogenase (6) | Clostridium acetobutylicum ATCC 824, Pseudomonas pseudoalcaligenes JS52, Pseudomonas aeruginosa Strain, Burkholderia sp. strain DNT. | 6 | ||
| 23 | hsaC; 3-hydroxy-9,10-secoandrosta-1,3,5(10)-triene-9,17-dione monooxygenase (7) | Mycobacterium tuberculosis CYP142, Comamonas testosterone | 7 | ||
| 24 | vgb; virginiamycin B lyase (5) | Staphylococcus aureus | 5 | ||
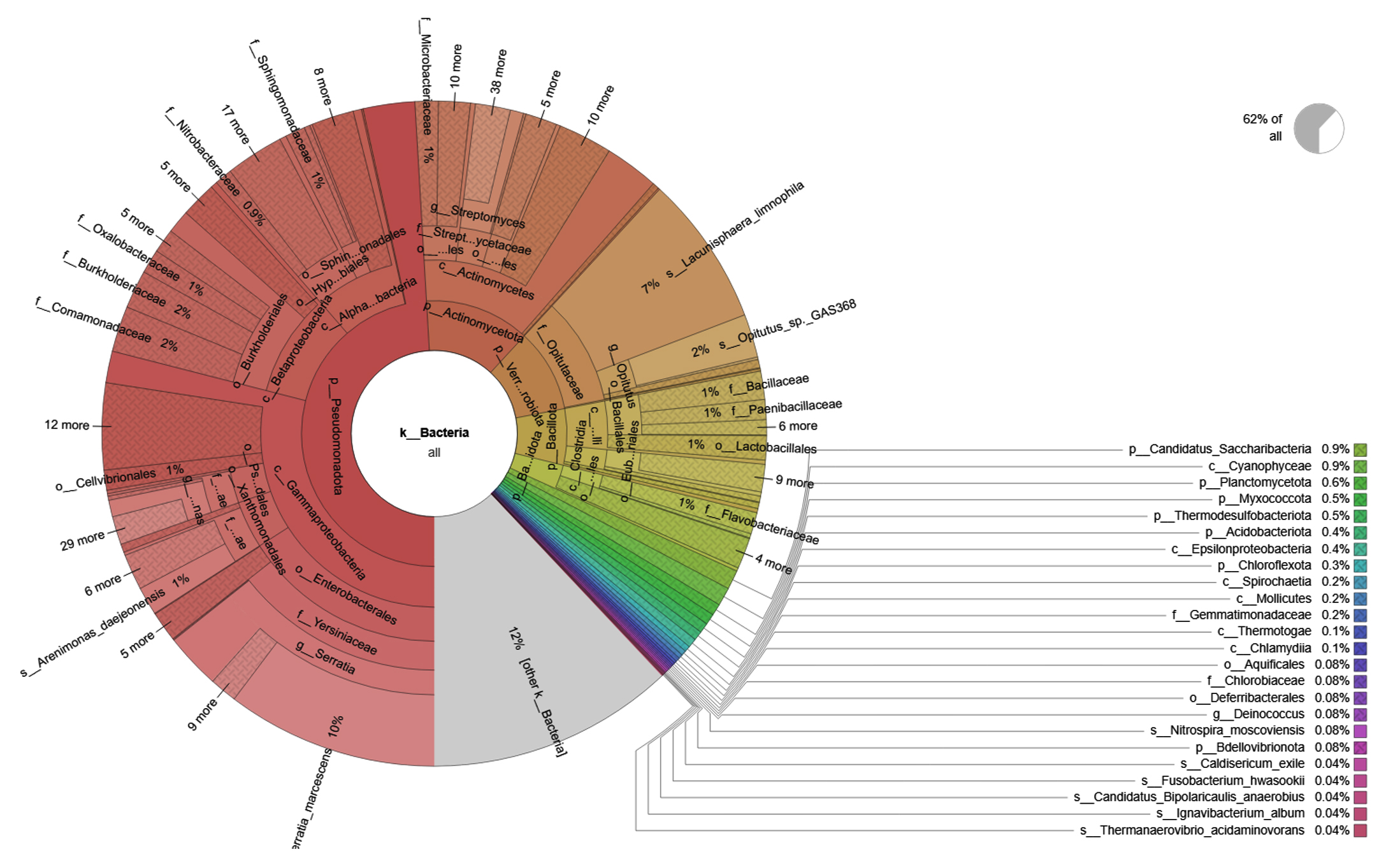
Figure 1. Krona Plot of SA4 sample represent metagenomic microbial communities
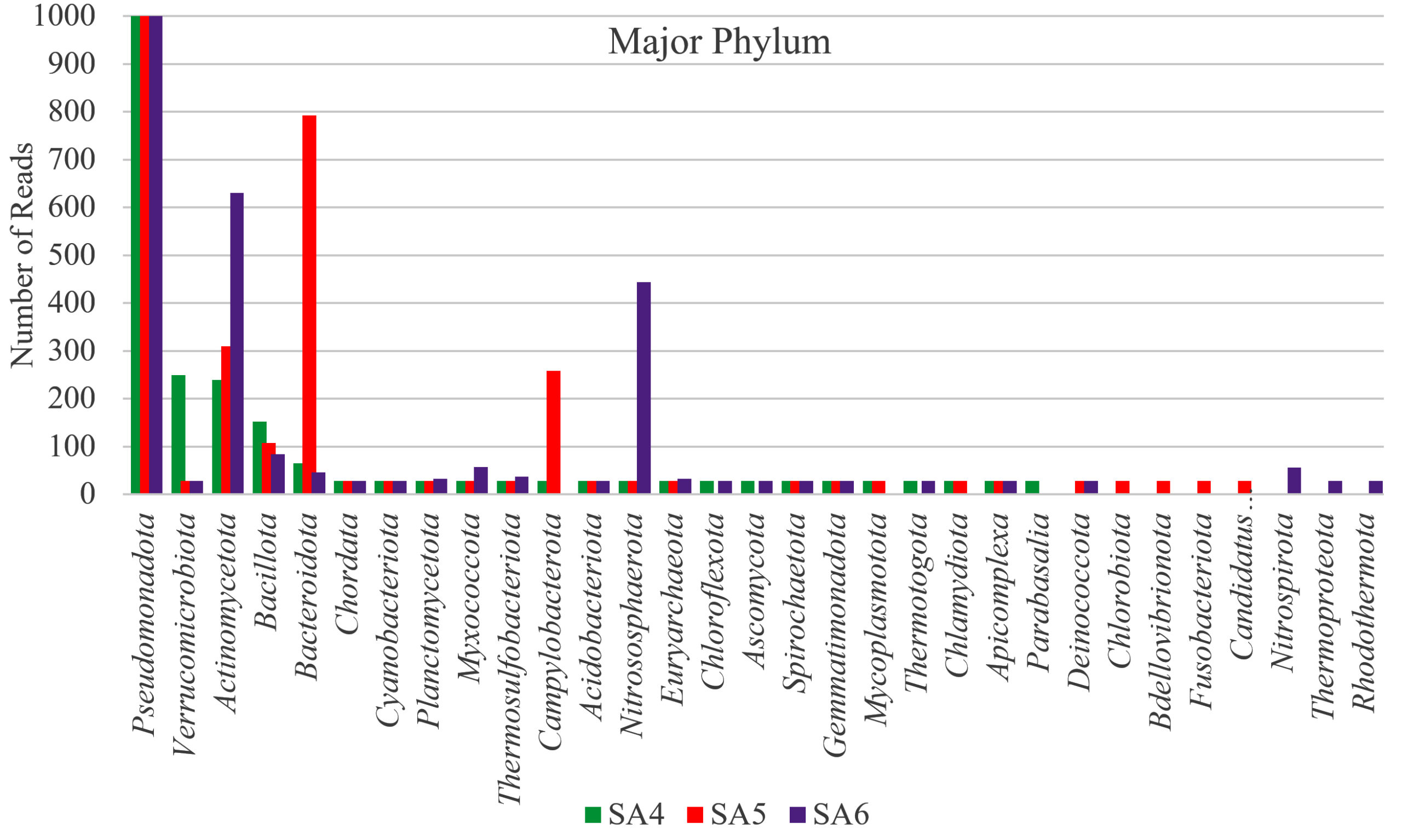
Figure 2. Graphical representation of Major Phylum dominating in SA4, SA5, and SA6 sample
Comparative microbial community analysis of Metagenomic DNA
Taxonomic analysis of metagenomic DNA reveals that the microbial community of SA4 primarily composed of four major phyla including Pseudomonadota, Actinomycetota, Verrucomicrobiota and Bacillota (Figure 1 and 2). Among these, Pseudomonadota is the most prevalent phylum, constituting (54.0%) of all identified phylotypes. Within this phylum, the dominant groups include g-Proteobacteria (59%) and β-proteobacteria (20%) and α-proteobacteria (16%). The combined proportion of Actinomycetota, Verrucomicrobiota and Bacillota account for 30% (13%, 10% and 7% respectively). Notably, each phylum is predominantly represented by one or two sub-groups, such as Yersiniaceae, Pseudomonadaceae and Xanthomonadales within the g-Proteobacteria, Burkholderiales, Nitrosomonadales and Rhodocyclales in β-Proteobacteria, Hyphomicrobiales, Sphingomonadales, Rhodobacterales and Rhodospirillales in α-Proteobacteria, Micrococcales and Streptomycetaceae in Actinomycetota, Opitutaceae in Verrucomicrobiota, Bacilli and Clostridia in Bacillota. The phylum having major dominating genus in SA4 sample was Serratia, Lacunisphaera and Opitutus (Figure 2 and 3). Similarly, taxonomic characterization of SA5 soil sample reveals microbial diversity having Pseudomonadota, Bacteroidota, Actinomycetota and Campylobacterota are the major phylum. Pseudomonadota was the highest amongst having (81%) of all phylotype, where γ- Proteobacteria (76%), α-Proteobacteria (13%) and β-Proteobacteria (8%) were the dominant parts. Simultaneously Bacteroidota, Actinomycetota and Campylobacterota making up 14% (8%, 4%, and 2%, respectively). Furthermore, SA6 soil sample was also dominated by Pseudomonadota, Actinomycetota, Nitrososphaerota, and Bacillota, amongst which Pseudomonadota is the highest phylum (62%) of all the phylotype, where β-Proteobacteria (40%) followed by α-Proteobacteria (28%), and γ–Proteobacteria (26%). Actinomycetota, Nitrososphaerota, and Bacillota represent 24% of bacterial diversity (18%, 4%, and 2%, respectively). These phyla were furtherly predominated by one of the following sub-groups which includes Xanthomonadaceae, Pseudomonadaceae, Sphingomonadaceae, Chromatiaceae, Flavobacteriaceae, Sulfurimonadaceae, Comamonadaceae, Nitrosopumilaceae, and Nitrobacteraceae etc. (Figure 2 and 3) (Figure S2 and S3). The strains of sub-groups have been found that they are actively playing crucial role in degradation of various xenobiotic compounds which includes petroleum and other aromatic and aliphatic hydrocarbons.25,51,52 A substantial diversity in the structure of microbiome is evident among samples derived from various contaminated and non-contaminated soil source. The dominance of microbial communities such as g-Proteobacteria, a-Proteobacteria, and Acidobacteriota were significantly different amongst soil contaminated with oil, non-contaminated and significantly varies amongst high salinity soils.25,53-55 This studies suggest that the microbial communities are highly diverse and intricately linked to soil compositions that underpin these ecosystems.56,57 Taxonomic analyses revealed that “Proteobacteria” and “Pseudomonas” were the major dominating phylum and genus within soil microbiome, respectively.10,58,59 Numerous studies have focused on various aspects, such as unveiling bacterial communities capable to fix nitrogen from petroleum contaminated soil, the detection of microbial communities from long term contaminated soil with different aromatic hydrocarbons such as benzoate, biphenyl, and naphthalene. The detection of microbial communities capable to bioremediation and biodegradation of soil contaminated with various hydrocarbons along with some toxic metals like chromium and arsenic.60-63 Furthermore, pyrosequencing of soil contaminated with crude oil within China and Russia unveiled the bacterial communities capable to utilize phenanthrene, have been conducted.64-66 These investigations have consistently suggested that the presence and abundance of Proteobacteria. The current study has unveiled distinctive microbial communities within specific niches of industrial effluent contaminated soils. The metagenomic analysis of this industrial effluent contaminated soil samples reveal different microbial species which involve in degradation of various xenobiotic compounds, some of the species like Sulfuricurvum kujiense, Novosphingobium sp., Variovorax paradoxus, Usitatibacter rugosus, Cupriavidus sp., and many others found in SA4, SA5, and SA6 soil samples. These investigation supports the previously reported many metagenomic studies.67
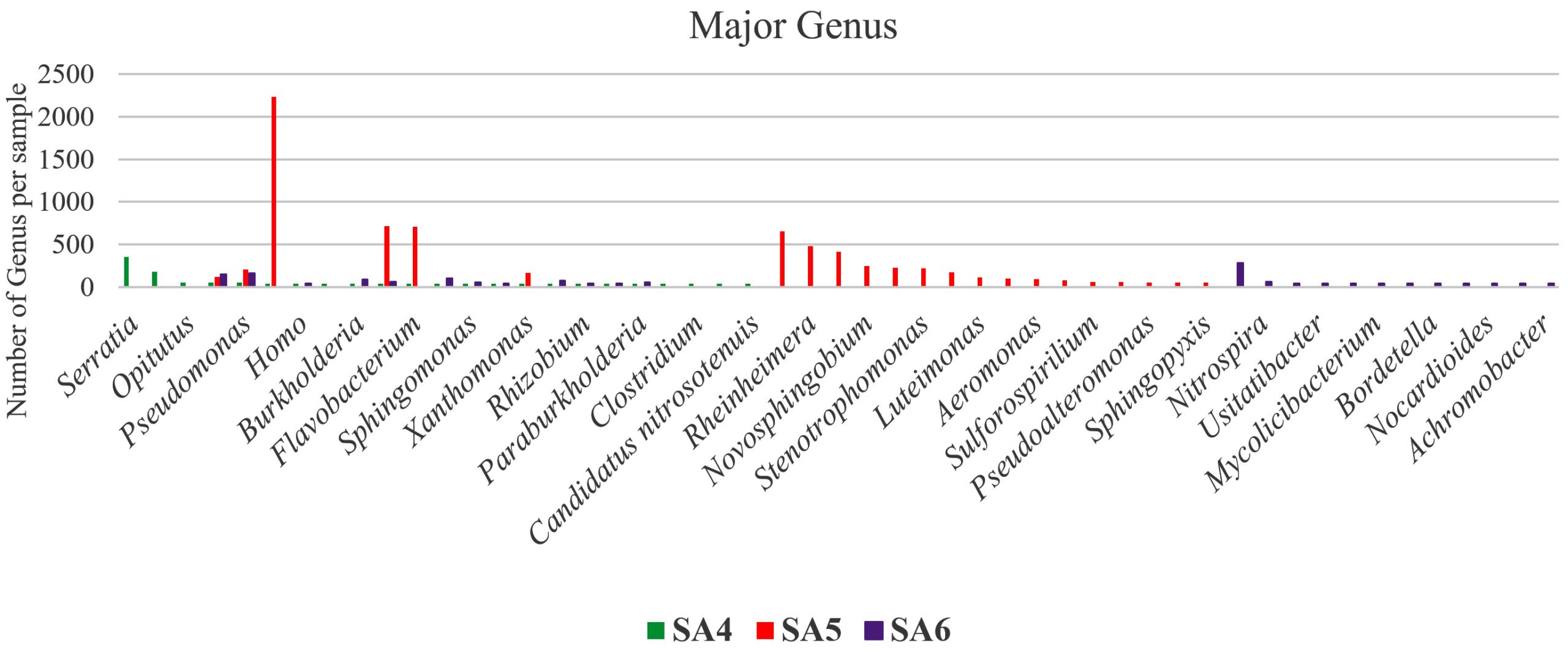
Figure 3. Graphical representation of major genus dominating in SA4, SA5, and SA6
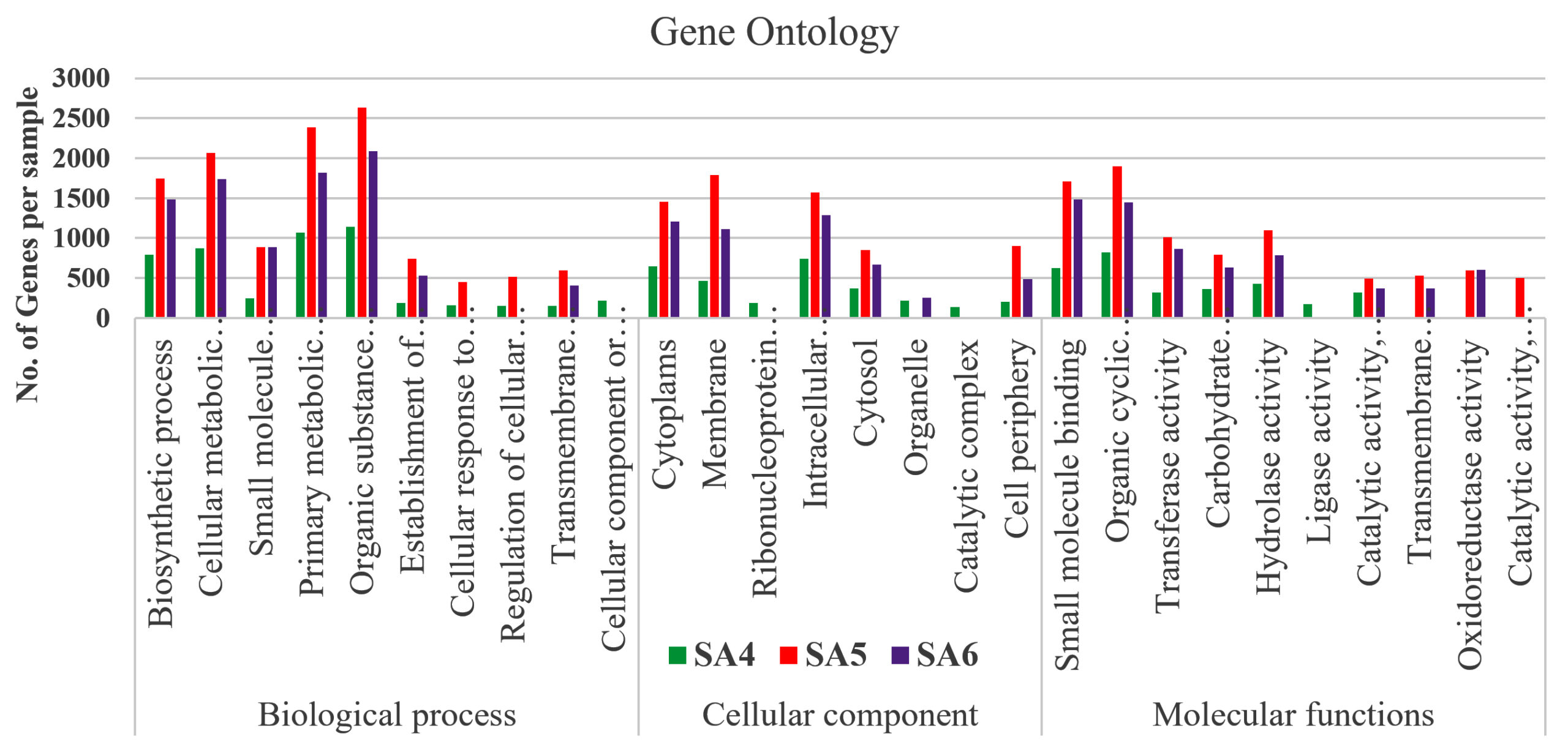
Figure 4. Graphical representation of Gene ontology of SA4, SA5, and SA6
Functional annotation and metabolic diversity insight metagenome
The microbial community of soil samples greatly varies with its metabolic diversity because of soil composition and microbial community structure. On analysis of metabolic versatility of the microorganisms present in our SA4, SA5, and SA6 sample by classifying on the basis of open reading frames (ORFs) followed by KEGG orthology groups (KOs) which furtherly used to analyze different metabolic pathways using KEGG and COG databases. Each pathway of these databases unveiled the different metabolic functions, pathways, enzymes, and reveals distinct patterns utilized by microbial communities present in contaminated soil (Figure 4). These pathways are categorized into three major groups. Group one consist of biological process having highly represented pathways, including organic substance metabolism, primary metabolic process, cellular metabolic process, biosynthetic process, small molecules metabolic process followed by some other transport system, as well as cellular response to stimulus and other pathways are dominated in all the three SA4, SA5, and SA6 samples.
The pathways represented in group one is essential for all living organisms which plays significant role in their physiological needs, which resulting their continuous representation amongst all group of organisms. Although group one pathways account for 34% of the open reading frames (ORFs). Similarly, the pathways in groups two, which is lowest in the rank, represents only 20% of the predicted open reading frames (ORFs), which includes major pathways for the cellular components like membrane synthesis, cytoplasm synthesis, intracellular anatomical process as well as for cytosol and cell periphery. The highly mosaic distribution of group three pathway suggests that these functions are specific to certain microbiomes. The remaining 31% of pathway categories, classified as group three, shows moderate representation, involving pathways responsible for all molecular functions which includes organic cyclic compounds binding, small molecule binding, transferase activity, some of are highly represent the hydrolase activity, oxidoreductase activity, catalytic activity, and ligases activity, which plays crucial role in cellular responses to environmental stresses such as degradation of aromatic and aliphatic hydrocarbon compounds (Figure 4). Similarly, the assembled contigs of SA4, SA5, and SA6 underwent analysis through the assignment of predicted functions to genes utilizing COGs. The functional annotation of COG genes was classified into different categories and represented by A-Z in Figure 5. Among these COGs representation the highest contigs were dominated by unknown functions (S), followed by “Translation (J), “Cell wall/membrane/ development and biogenesis (M), “ Replication and repair (L)”. The major class designated as “Amino acid metabolism and transport (E) and “Energy production and conservation (C)” was significantly represented for its metabolic activities like oxidoreductases activity, which was also reported for the degradation of azo dyes and xenobiotic compounds such as aromatic hydrocarbons under stress conditions.68 Additionally, other categories such as “Nucleotide metabolisms and transport (F)”, “Carbohydrate metabolism and transport (G)”, “Coenzyme metabolism (H), “Lipid metabolism (I)”, “Transcription (K)”, “Post translation and modification (O)”, “Inorganic ion transport and metabolism (P)”, “Secondary metabolites biosynthesis, and co-metabolism (Q)”, “signal transduction mechanism (T)”, “Intracellular trafficking and secretion (U)”, and some of the genes are functionally active in “Defense mechanism (V)”. All these gene were directly or indirectly linked with transportation of ions and compounds which helps in various metabolic processes transport of ions and compounds, as well as various metabolic processes.
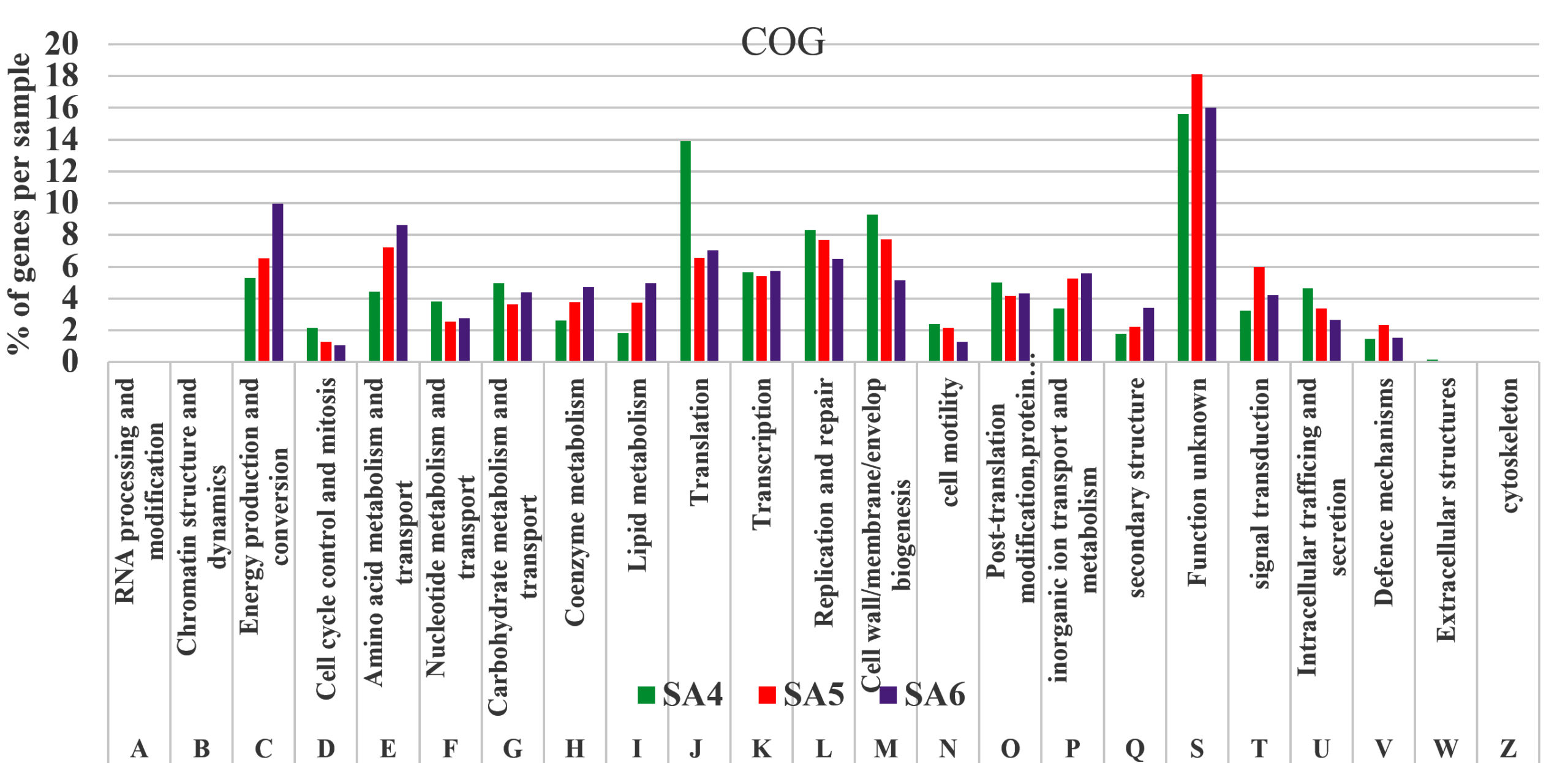
Figure 5. Graphical representation of COG of SA4, SA5, and SA6
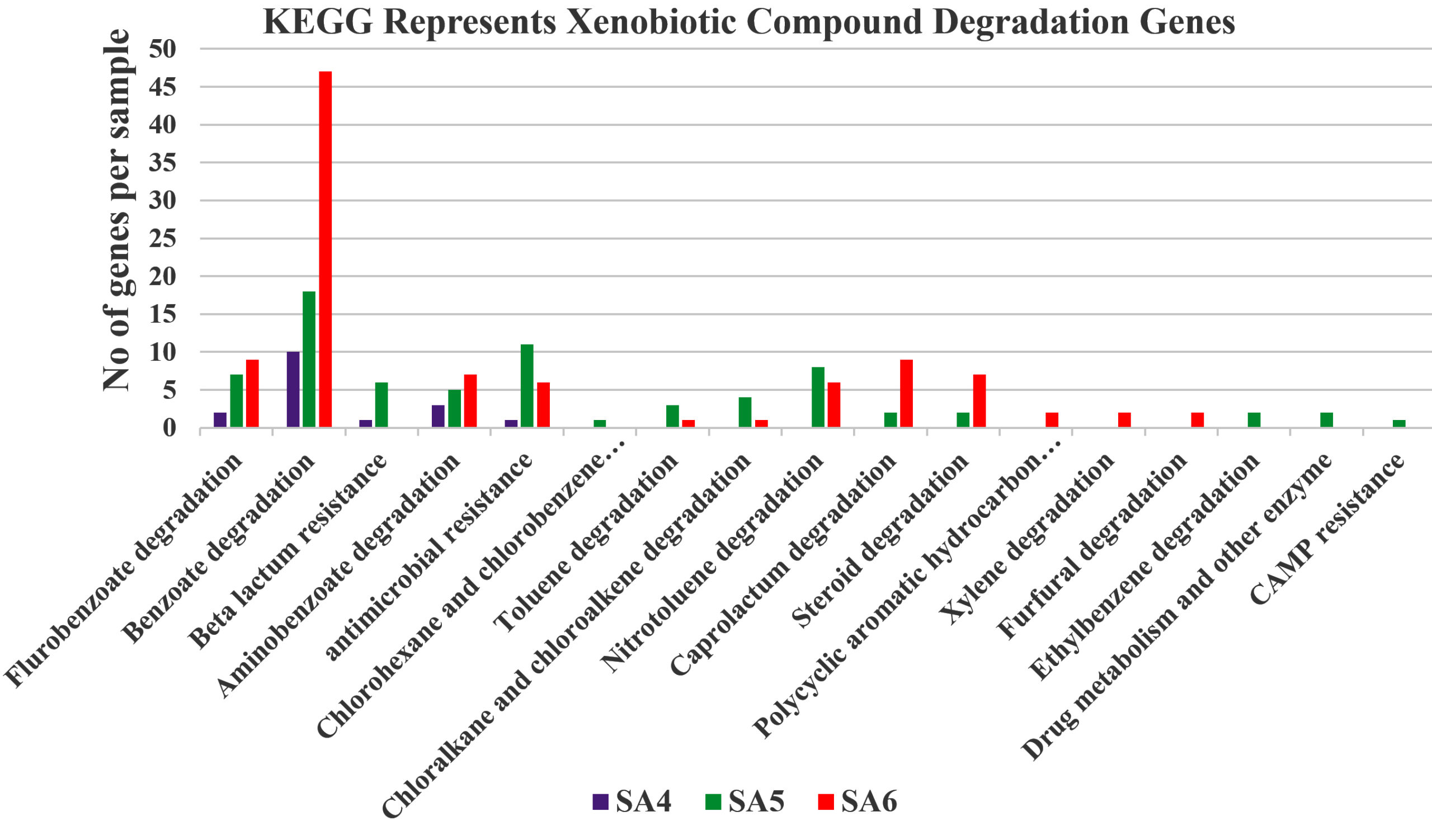
Figure 6. Graphical representation of KEGG represents xenobiotics compound degradation genes present in SA4, SA5, and SA6
On analysis of metagenomic data of bacterial genomes indicates the presence of microbial communities in industrial effluent contaminated soil samples having different xenobiotic compounds have capability to degrade such hydrocarbons and plays significant role in bioremediation processes. Numerous studies have elucidated that Pseudomonas stutzeri have great mechanisms to degrade and bioremediation capabilities of some crucial xenobiotic compounds like chlorobenzene, carbon tetrachloride, phenanthrene, toluene, xylene, and various azo dyes in effluents.25,62,69 Furthermore, genomic analyses of diverse Pseudomonas species have uncovered insights into the functions of oxygenase, oxidoreductases, ferredoxins, cytochromes, dehydrogenases, and proteins involved in sulfur metabolism.67,70 This genus is also characterized by numerous operons that facilitates the catabolism of wide array of aromatic compounds, as well as gene clusters that encode enzymes likely involved in metabolism of anthropogenic substrates. Additionally, Shewanella baltica is recognized for its significant contributions for degradation of different aromatic and aliphatic hydrocarbons which includes some of organic and inorganic compounds, including naphthalene and styrene, as well as under anaerobic condition this species were capable to immobilized and degrade some of toxic heavy metals including radionuclides.71,72 Some other studies reported genus like Rhodobacter have capability to decolorization of nitrophenol, chlorobenzenes, and azo dyes.25,54,73 In recent years, enzymes associated with environmental bioremediation have garnered substantial attention, leading to the implementation of various innovative approaches for in-depth studies of specific enzyme classes. It is observed that numbers of enzymes involved in degradations pathways were directly or indirectly linked through their proteomic rather than their taxonomic classification, which indicates that the genes involved in catabolic process have highly diverse mechanism and different processes.65 Some studies reported that the presence of tmo-like genes in Pseudomonas, Mycobacterium and Bradyrhizobium which are highly diverse with respect to phylogenetics, further supports the notion of horizontal gene transfer.52,59,74,75 To rightly assess the catabolic eventuality for biodegradation, it’s necessary to consider the wide range of catabolic pathways generated by microorganisms, as well as the diversity of enzymes within a single gene family or across multitudinous gene families. As a result, the exploration of catabolic gene eventuality must be done independently, as depending simply on taxonomic lives might leads to equivocal finding about functional assignments and affects in particular and undetermined relationship.
Identification of xenobiotic compound degradation pathways through KEGG
For the identification of xenobiotic aromatic and aliphatic hydrocarbon degradation pathways, the current metabolic pathways were compared with reference databases. The analysis indicates that the microbial communities of SA4, SA5, and SA6 exhibits an enrichment in degradation pathways for wide range of xenobiotic hydrocarbons, which includes toluene, xylene, fluorobenzoate, benzoate, aminobenzoate, chlorohexane and chlorobenzene, chloroalkane and chloroalkene, nitrotoluene, caprolactam, polyaromatic hydrocarbons, furfural and ethylbenzene. Some of other like beta-lactam resistance, antimicrobial resistance, drug metabolism and other enzyme, steroid degradation and CAMP resistance gene as well (Figure 6). These results highlight the metabolic capabilities of the current metagenomic community concerning xenobiotic compounds. The xenobiotic aromatic and aliphatic hydrocarbons are primarily consisting of alkyl, sulphonates, cycloalkanes, methoxy, nitro, halogen (like chlorine, bromine, fluorine etc.) or amino substituents of monocyclic aromatics (which including toluene, xylene, benzoate, aminobenzoate, flurobenzoate, nitrotoluene, ethylbenzene etc.) and bicyclic aromatic and aliphatic, heterocyclic compounds (such as bisphenol, chloroalkane and chloroalkene, polycyclic aromatic, furfural, PTA, and tetrachloroethene etc.). The findings align with the chemical characteristic of the soil samples of SA4, SA5, and SA6, which is also favors the studies data. The over-representation of pathways for degradation of xenobiotics indicates that metagenomic community were adaptable in utilizing these aromatic hydrocarbons as source of carbon and energy. Studies suggest that this microbial communities significantly utilize glycolysis or gluconeogenesis pathways, which indicates that these metagenomic communities not only utilize aromatic and aliphatic hydrocarbons as source of carbon and energy, but they may utilize glucose as primary source of carbon and energy (Figure 6).25
Subsequently the EC numbers were aligned with a curated list of enzymes responsible for biodegradation of xenobiotic compounds, as provided in the UM-BBD database. Such enzymes are E3.1.1.45; Carboxymethylenebutenolidase, Catechol 2,3-dioxygenase, Metallo-beta-lactamase class B, E1.14.13.40; Anthraniloyl-CoA monooxygenase, Aminoglycoside 6-adenylyltransferase, Protocatechuate 3,4-dioxygenase, Toluene monooxygenase, E3.8.1.2; 2-Haloacid dehalogenase, N-ethylmaleimide reductase, 2-Hydroxy-6-oxo-octa-2,4-dienoate hydrolase, Cyclohexanone monooxygenase, Thiopurine S-methyltransferase, 3-Oxosteroid 1-dehydrogenase, Chloramphenicol O-acetyltransferase, 4-Carboxymuconolactone decarboxylase, Benzoyl-CoA reductase, 4-Hydroxybenzoyl-CoA reductase, 2,5-Furandicarboxylate decarboxylase, p-cumate 2,3-dioxygenase, pht3; phthalate 4,5-dioxygenase, Haloacetate dehalogenase, Hydrogenase, 3-Hydroxy-9,10-secoandrosta-1,3,5(10)-triene-9,17-dione monooxygenase, Virginiamycin B lyase, and many others plays significant role in biodegradation of various aromatic and aliphatic hydrocarbons and some of were also involve in antimicrobial resistance as well Table. Further in-depth investigation is required to explore the preferential utilization of these source. The microbial processes critical to biodegradation, including oxidoreduction process, binding, immobilizing, and biotransformation, are facilitated by various enzymes such as oxidoreductases, and oxygenase’s.69,76 Nonetheless, only a limited number of specific enzymes directly participate in biodegradation. In contrast, numerous enzymes, which primarily function in cellular metabolic processes, can assume alternative roles in biodegradation pathways which exposed to stressors like hydrocarbons, dyes, and xenobiotic compounds. Many of these enzymes exhibits promiscuous activities.44,77 The concept of “catalytic promiscuity” refers to an enzyme’s ability to catalyze multiple reactions, termed secondary activities, at its active site. From a fundamental perspective, investigation into catalytic promiscuity provide insights into the evolutionary development of enzymes and facilitate the adaptation of enzymes for specific application in vitro. To assess the genetic capabilities of these bacterial communities in industrial effluent contaminated soil and their adaptive mechanisms for the biodegradation of xenobiotic substances, various methods for functional annotation and gene characterization have been employed. Metagenomic single reads and contigs have been annotated using Gene Ontology (GO) terms, Clusters of Orthologous Group (COG) accessions, and Kyoto Encyclopedia of Gene and Genome (KEGG) identifiers, followed by assigning these predicted functional genes to coding sequences. The anticipated genes and enzymes were integrated into metabolic pathways to clarify the potential catabolic routes utilized by the resident microbial community.78
Numerous additional studies have been conducted to elucidate the microbial metabolism associated with xenobiotic biodegradation. For instance,79 presented a comprehensive sequential degradation pathways for conversion of polycyclic aromatic hydrocarbon in to β-ketoadipate pathway via protocatechuate, ultimately resulting in mineralization to carbon dioxide through the tricarboxylic acid cycle.54 In the review of Perez-Pantoja et al.80 elucidate the metabolic capabilities of Cupriavidus necator JMP134 having capability of bioremediate different xenobiotic hydrocarbons. Out of 140 aromatic compounds evaluated, 60 were utilized as exclusive source of carbon an energy, which aligns closely with the catabolic functions inferred from genomic analysis. C. necator possesses nearly all principal ring-cleavage pathways for degradation of aromatic hydrocarbons, including the b-ketoadipate pathway (which follows enzyme-mediated aryl-ring degradation), these pathways encompasses branches for catechol, chlorocatechol, methyl catechol, and protocatechuate ortho and meta ring-cleavage pathway; catechol ortho and meta ring cleavage pathway; quinoline pathway; b-carboxy-cis, cis-muconate pathway; shikimate pathway; gentisate pathway; phenylacetyl-CoA pathway; 2-aminobenzoyl-CoA pathway; benzoyl-CoA pathway; 3-hydroxyanthranilate pathway and many others.61 The presence of C. necator in our metagenomic reads further supports the potential for these aforementioned metabolic capabilities.
A comprehensive metabolic pathway proposed for the degradation of xenobiotic hydrocarbon degradation
The hypothetical comprehensive metabolic pathway has been constructed for the degradation of aromatic and aliphatic hydrocarbons based on the interaction among the degradation pathways identified in the studied soil metagenome of SA4, SA5, and SA6 (Figure 7). The thorough examination of these pathways and enzymes associated with degradation of xenobiotic compounds indicates that the biodegradation of these compounds undergoes through aerobic and anaerobic conditions as follows: the toluene and xylene degradation initiated with hydroxylation reaction to form intermediates of toluene and xylene are methylcatechol via enzyme (EC 1.14.13.2). This intermediates furtherly oxidized into catechol which undergoes ring cleavage by dioxygenases (EC 1.13.11.1 and EC 1.13.11.2) which is again breakdown into Maley acetate via (EC 1.3.2.32) which is furtherly converted into succinyl CoA and Acetyl CoA. Similarly, aminobenzoate is derivative of benzoate containing amino group. The degradation of these compounds involves hydroxylation of aminobenzoate to form hydroxyaminobenzoate via (EC 1.14.12.1) which is furtherly converted into catechol by (EC 1.13.11.1 and EC 1.13.11.2). This catechol undergoes oxidation to produce intermediates like succinyl CoA and acetyl CoA. The degradation of chlorinated compounds initiated by (EC 1.14.12.3 and EC 1.14.12.26), it requires initially dichlorination which is done by (EC 1.21.99.5) result into formation of chlorocyclohexane and chlorobenzene which is furtherly undergoes hydroxylation to form meleyacetate. This intermediate is furtherly breakdown to form intermediates of TCA cycles. Similarly, the degradation of flurobenzoate initiated with hydroxylation and oxidation, which result into formation of 4-hydroxybenzoate by (EC 1.14.12.10 and EC 1.14.12.1). It is furtherly entered into benzoate degradation pathway; were it get oxidized by (EC 1.14.13.2) to form intermediates like succinyl CoA and acetyl CoA. Similarly, the polycyclic aromatic hydrocarbon degradation pathway is initiated with oxidation of anthracene, fluorene via monooxygenase (EC 1.14.12.12), and phenanthrene and pyrene via (EC 1.13.11.- and EC 1.14.-.-), respectively. This result into hydroxylation of anthracene to form 1-hydroxyanthracene. This intermediates furtherly oxidized to form catechol via (EC 1.4.13.1) which enter into benzoate degradation pathway; were it get oxidized by dioxygenases like (EC 1.13.11.1 and EC 1. 13.11.2) to form intermediates of TCA cycle like succinyl CoA and acetyl CoA, each of these intermediates are enter into TCA cycle for energy conservation (Figure 7).
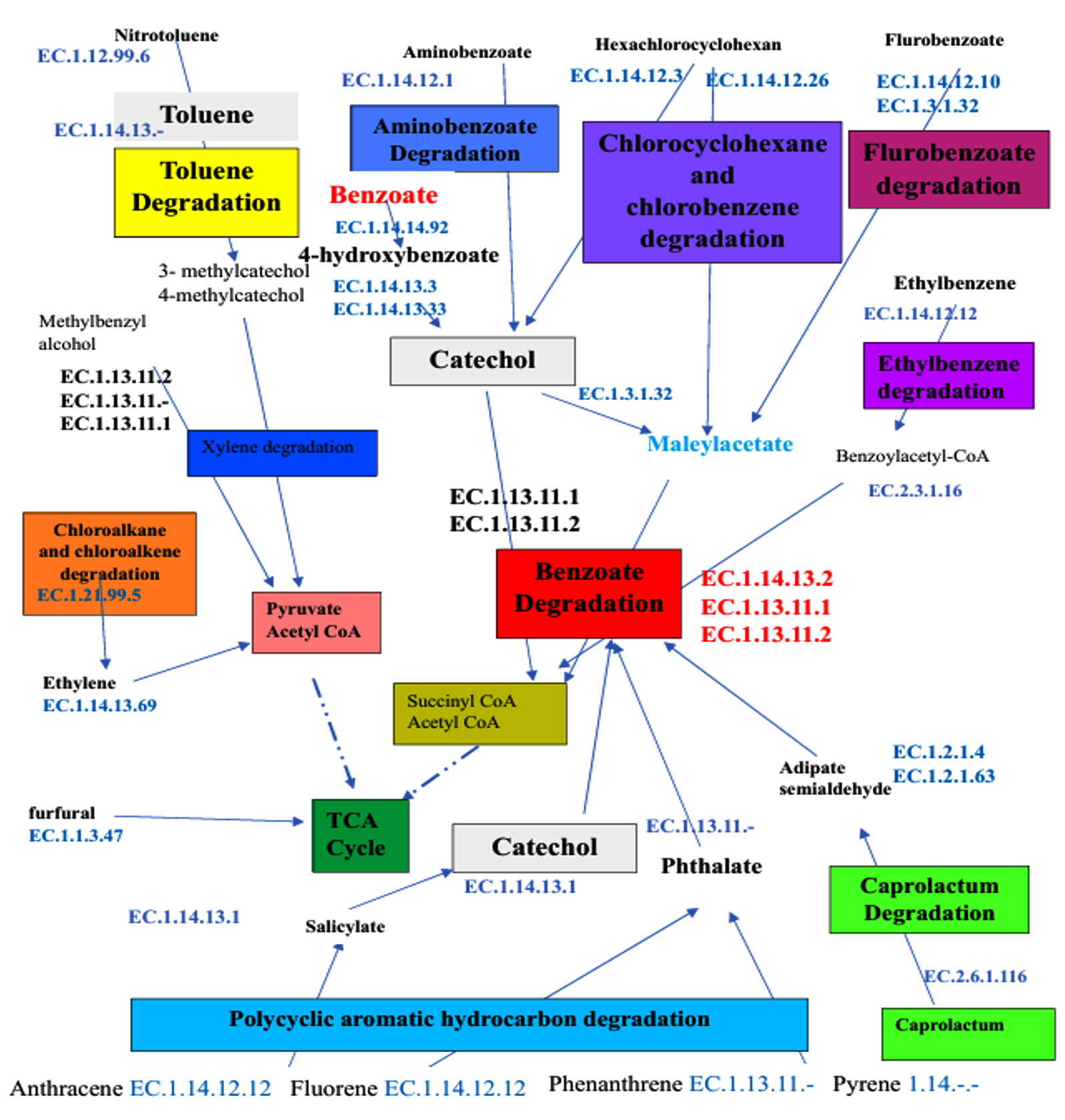
Figure 7. Overall schematic representation of metabolic pathways responsible for degradation of various aliphatic and aromatic hydrocarbon of SA4, SA5, and SA6
The significant prevalence of degradation pathways for a wide array of such aromatic and aliphatic hydrocarbon compounds underscores a remarkable anabolic and catabolic capabilities of the current microbial communities present in this industrial effluent contaminated soil samples. Such level of diversity has been reported by Bao et al.25 for metagenomic investigation of hydrocarbon contaminated soils.64 reported genes and enzymes like catechol dioxygenase responsible for degradation of hydrocarbons via high throughput metagenomic sequencing of soil tainted by jet fuel. Similarly, Eze and Lee et al. 81,82 reported microbial communities responsible for the degradation of various hydrocarbons like benzoate, toluene, ethylbenzene and xylene under anaerobic conditions. They also reported that, biodegradation of this compounds under anaerobic conditions leads to accumulation of significant levels of uncleaved aromatic acids (like benzoic acids, phenolic acids and phthalic acids) which may helpful for the breakdown of some other phenolic compounds.83 Similarly, some other findings associated with the degradation of BTEX include benzyl alcohol,84,85 benzoic acids, propionic acid, hydroxy acetophenone, and dimethyl benzaldehyde. These metabolites were identified during degradation process. In another study of Andreote et al.86 uses high throughput metagenomic sequencing using 454 sequencing technologies to investigate the microbial communities of soil contaminated with oil, and reported that the genes function were limited to metabolism of methane, formaldehyde and CO2. While another study of Yergeau et al.87 reported that presence of high numbers of genes responsible for degradation of aromatic and aliphatic hydrocarbon degradation using 454 sequencing technology to analyze the metagenome of diesel-polluted soils, still there is no enriched pathways is identified for the degradation of such xenobiotics.
Our findings indicates that the metagenome of industrial effluent contaminated soil samples having significant level of diversity in microbial communities and their metabolic processes. On the basis of comparative studies and detailed analysis it is found that microbial communities are responsible for biodegradation of different aromatic and aliphatic hydrocarbons such as toluene, xylene, chlorobenzene, aminobenzoate, benzoate, nitrotoluene, chloroalkane and chloroalkene, cyclochlorohexane, ethylbenzene, caprolactam, polycyclic aromatic hydrocarbons, flurobenzoate, furfural, and other aromatic hydrocarbons. Furthermore, a comprehensive hypothetical metabolic pathway was constructed on the basis of overall metabolic pathways identified for the degradation of xenobiotic compounds. The insight gained from this research will be instrumental in developing effective bioremediation processes to remediate such industrially effluent contaminated soils.
Additional file: Additional Table S1-S4 and Figure S1-S3.
ACKNOWLEDGMENTS
The authors would like to thank Ministry of Tribal Affairs for providing National Fellowship Scheme for ST Students, and Eurofins Genomics India Pvt. Ltd., Karnataka, India, for their help in NGS Metagenomic sequencing analysis of all the soil samples. Authors are also thankful to Dr. Prof. Narayana Kamath (NAMO Medical Education and Research Institute, Silvassa, D & N.H) for his support and help in providing Molecular Laboratory for analysis of soil samples.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
AUTHORS’ CONTRIBUTION
NB conceptualized the study, collected resources and applied methodology. NB performed visualization, Investigation and formal analysis. AJT performed data curation and validation. MD performed supervision and project administration. NB wrote the original draft. NB, MD and AJT reviewed the manuscript. AJT wrote the manuscript. NB and AJT edited the manuscript. All authors read and approved the final manuscript for publication.
FUNDING
This study was supported through fellowship provided by Ministry of Tribal Affairs, National Fellowship Scheme for ST Students.
DATA AVAILABILITY
The whole metagenomic sequence data of these 3 samples were submitted in NCBI databases under the NCBI Bioproject number PRJNA1084772. The metagenomic sequencing data is available in NCBI Sequence Read Archive under accession numbers SAMN46715193 (SA4 soil sample), SAMN46715194 (SA5 soil sample) and SAMN46715195 (SA6 soil sample).
ETHICS STATEMENT
Not applicable.
- Pei Y, Tao C, Ling Z, et al. Exploring novel Cr(VI) remediation genes for Cr(VI)-contaminated industrial wastewater treatment by comparative metatranscriptomics and metagenomics. Sci Total Environ. 2020;742:140435.
Crossref - Ghosh A, Mehta A, Khan AM. Metagenomic analysis and its applications. Encycl Bioinforma Comput Biol ABC Bioinforma. 2019;3:184-193.
Crossref - Zhang L, Calvo-Bado L, Murray AK, et al. Novel clinically relevant antibiotic resistance genes associated with sewage sludge and industrial waste streams revealed by functional metagenomic screening. Environ Int. 2019;132:105120.
Crossref - Rambo IM, Dombrowski N, Constant L, Erdner D, Baker BJ. Metabolic relationships of uncultured bacteria associated with the microalgae Gambierdiscus. 2020;22(5):1764-1783.
Crossref - Zhang L, Loh KC, Lim JW, Zhang J. Bioinformatics analysis of metagenomics data of biogas-producing microbial communities in anaerobic digesters: A review. Renew Sustain Energy Rev. 2019;100:110-126.
Crossref - Zhang L, Lv J. Metagenomic analysis of microbial community and function reveals the response of soil respiration to the conversion of cropland to plantations in the Loess Plateau of China. Glob Ecol Conserv. 2020;23(10):e01067.
Crossref - Nannipieri P, Ascher J, Ceccherini MT, Landi L, Pietramellara G, Renella G. Microbial diversity and soil functions. Eur J Soil Sci. 2003;54(4):655-670.
Crossref - Kumar A, Tripti, Raj D, Maiti SK, Maleva M, Borisova G. Soil Pollution and Plant Efficiency Indices for Phytoremediation of Heavy Metal(loid)s: Two-Decade Study (2002–2021). Metals. 2022;12(8):1330.
Crossref - Hugenholtz P. Exploring prokaryotic diversity in the genomic era. Genome biol. 2002;3:1-8.
Crossref - Lazaro-Mass S, Gomez-Cornelio S, Castillo-Vidal M, Alvarez-Villagomez CS, Quintana P, De la Rosa-Garcia S. Biodegradation of hydrocarbons from contaminated soils by microbial consortia: A laboratory microcosm study. Electron J Biotechnol. 2023;61:24-32.
Crossref - Bastida F, Garcia C, Fierer N, et al. Global ecological predictors of the soil priming effect. Nat Commun. 2019;10(1):3481.
Crossref - Yu Z, Liang K, Huang G, et al. Soil Bacterial Community Shifts Are Driven by Soil Nutrient Availability along a Teak Plantation Chronosequence in Tropical Forests in China. Biology. 2021;10(12):1329.
Crossref - Landrigan PJ, Fuller R, Acosta NJR, et al. The Lancet Commission on pollution and health. Lancet. 2018;391(10119):462-512.
Crossref - Kurniawan T, Anouzla A (eds). Handbook of Microplastic Pollution in the Environment: Microplastic Pollution in the soil. CRC Press. 2025.
Crossref - Schloter M, Nannipieri P, Sorensen SJ, van Elsas JD. Microbial indicators for soil quality. Biol Fertil Soils. 2018;54(1):1-10.
Crossref - Li X, Zhang Y, Gulbins E. Handbook of Hydrocarbon and Lipid Microbiology. Handb Hydrocarb Lipid Microbiol. 2010.
Crossref - Yin S, Zhang X, Xie J, Ye X, Zhang X. Change of microbial communities in heavy metals-contaminated rhizosphere soil with ectomycorrhizal fungi Suillus luteus inoculation. Appl Soil Ecol. 2023;190:105019.
Crossref - Li Q, You P, Hu Q, et al. Effects of co-contamination of heavy metals and total petroleum hydrocarbons on soil bacterial community and function network reconstitution. Ecotoxicol Environ Saf. 2020;204:111083.
Crossref - Sharma P, Kumar S, Pandey A. Bioremediated techniques for remediation of metal pollutants using metagenomics approaches: A review. J Environ Chem Eng. 2021;9(4):105684.
Crossref - Zhao C, Zhang W, Guo Y, et al. Flocculent sludge outperforms filler biofilm for high salinity oilfield produced water treatment: Performance, metabolic pathways, and microbial communities. J Hazard Mater. 2025;492:138217.
Crossref - Wang M, Zhao S, Wang L, et al. Salt stress-induced changes in microbial community structures and metabolic processes result in increased soil cadmium availability. Sci Total Environ. 2021;782(12):147125.
Crossref - Iverson V, Morris RM, Frazar CD, Berthiaume CT, Morales RL, Armbrust EV. Untangling Genomes from Metagenomes: Revealing an Uncultured Class of Marine Euryarchaeota. Science. 2012;335(6068):587-590.
Crossref - Cai Y, Chen H, Yuan R, Wang F, Chen Z, Zhou B. Metagenomic analysis of soil microbial community under PFOA and PFOS stress. Environ Res. 2020;188:109838.
Crossref - Mishra S, Lin Z, Pang S, Zhang W, Bhatt P, Chen S. Recent Advanced Technologies for the Characterization of Xenobiotic-Degrading Microorganisms and Microbial Communities. Front Bioeng Biotechnol. 2021;9:632059.
Crossref - Bao YJ, Xu Z, Li Y, Yao Z, Sun J, Song H. High-throughput metagenomic analysis of petroleum-contaminated soil microbiome reveals the versatility in xenobiotic aromatics metabolism. J Environ Sci. 2017;56:25-35.
Crossref - Oduro D, Darko S, Blankson ER, Mensah GI. Assessment of Bacteria Contaminants in Different Zones and Point Sources of Sandy Beaches in Accra, Ghana. Microbiol Insights. 2023;16:11786361231195152.
Crossref - dos Santos OAQ, da Silva ENC, Garcia AC, et al. Impact of land use on Histosols properties in urban agriculture ecosystems of Rio de Janeiro, Brazil. Rev Bras Ciencia do Solo. 2020;44.
Crossref - Daniel C. Harris. Quantitative Chemical Analysis. Vol 5. 7th ed. (Mamontov VA, ed.). Craig Bleyer. 1971.
Crossref - Wilfinger WW, Mackey K, Chomczynski P. Effect of pH and Ionic Strength on the Spectrophotometric Assessment of Nucleic Acid Purity. Biotechniques. 1997;22(3):474-481.
Crossref - Zhou J, Bruns MA, Tiedje JM. DNA recovery from soils of diverse composition. Appl Environ Microbiol. 1996;62(2):316-322.
Crossref - Senes-Guerrero C, Gimenez S, Pacheco A, Gradilla-Hernandez MS, Schubler A. New MiSeq based strategy exposed plant-preferential arbuscular mycorrhizal fungal communities in arid soils of Mexico. Symbiosis. 2020;81(3):235-246.
Crossref - Bolyen E, Rideout JR, Dillon MR, et al. QIIME 2: Reproducible, interactive, scalable, and extensible microbiome data science. Peer J Preprints. 2018.
Crossref - Li D, Liu CM, Luo R, Sadakane K, Lam TW. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics. 2015;31(10):1674-1676.
Crossref - Chen S, Zhou Y, Chen Y, Gu J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34(17):i884-i890.
Crossref - Quast C, Pruesse E, Yilmaz P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2012;41(D1):D590-D596.
Crossref - Kassambara A. rstatix: Pipe-Friendly Framework for Basic Statistical Tests. CRAN Contrib Packag. 2019.
Crossref - Wood DE, Lu J, Langmead B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019;20(1):257.
Crossref - Cantalapiedra CP, Hernandez-Plaza A, Letunic I, Bork P, Huerta-Cepas J. eggNOG-mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Tamura K, ed. Mol Biol Evol. 2021;38(12):5825-5829.
Crossref - Klukas C, Schreiber F. Dynamic exploration and editing of KEGG pathway diagrams. Bioinformatics. 2007;23(3):344-350.
Crossref - Kanehisa M, Goto S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000;28(1):27-30.
Crossref - Lu C, Hong Y, Liu J, et al. A PAH-degrading bacterial community enriched with contaminated agricultural soil and its utility for microbial bioremediation. Environ Pollut. 2019;251:773-782.
Crossref - Meckenstock RU, Boll M, Mouttaki H, et al. Anaerobic degradation of benzene and polycyclic aromatic hydrocarbons. J Mol Microbiol Biotechnol. 2016;26(1-3):92-118.
Crossref - Weiland-Brauer N, Fischer MA, Schramm KW, Schmitz RA. Polychlorinated Biphenyl (PCB)-Degrading Potential of Microbes Present in a Cryoconite of Jamtalferner Glacier. Front Microbiol. 2017;8:1105.
Crossref - Sandhu M, Paul AT, Jha PN. Metagenomic analysis for taxonomic and functional potential of Polyaromatic hydrocarbons (PAHs) and Polychlorinated biphenyl (PCB) degrading bacterial communities in steel industrial soil. Poretsky RS, ed. PLoS One. 2022;17(4):e0266808.
Crossref - Nicolitch O, Feucherolles M, Churin JL, Fauchery L, Turpault MP, Uroz S. A microcosm approach highlights the response of soil mineral weathering bacterial communities to an increase of K and Mg availability. Sci Rep. 2019;9(1):14403.
Crossref - Bano S, Pervez S, Chow JC, et al. Coarse particle (PM10-2.5) source profiles for emissions from domestic cooking and industrial process in Central India. Sci Total Environ. 2018;627:1137-1145.
Crossref - Uritskiy GV. Microbial Community Adaptations and Dynamics in Extremophile Microbiomes of the Atacama Desert. Front Microbiol. 2020;10:3160. https://jscholarship.library.jhu.edu/handle/1774.2/63449
- Zhu W, Lomsadze A, Borodovsky M. Ab initio gene identification in metagenomic sequences. Nucleic Acids Res. 2010;38(12):e132-e132.
Crossref - Valderrama JA, Durante-Rodriguez G, Blazquez B, Garcםa JL, Carmona M, Diaz E. Bacterial degradation of benzoate: cross-regulation between aerobic and anaerobic pathways. J Biol Chem. 2012;287(13):10494-10508.
Crossref - Isaac P, Martinez FL, Bourguignon N, Sanchez LA, Ferrero MA. Improved PAHs removal performance by a defined bacterial consortium of indigenous Pseudomonas and actinobacteria from Patagonia, Argentina. Int Biodeterior Biodegrad. 2015;101:23-31.
Crossref - Shekhar SK, Godheja J, Modi DR. Molecular Technologies for Assessment of Bioremediation and Characterization of Microbial Communities at Pollutant-Contaminated Sites. In: Bharagava, R., Saxena, G. (eds) Bioremediation of Industrial Waste for Environmental Safety. Springer, Singapore. 2020:437-474.
Crossref - Singh N, Singh V, Rai SN, Vamanu E, Singh MP. Metagenomic Analysis of Garden Soil-Derived Microbial Consortia and Unveiling Their Metabolic Potential in Mitigating Toxic Hexavalent Chromium. Life. 2022;12(12):2094.
Crossref - Sun S, Wang Y, Xu C, et al. Reconstruction of microbiome and functionality accelerated crude oil biodegradation of 2,4-DCP-oil-contaminated soil systems using composite microbial agent B-Cl. J Hazard Mater. 2023;447:130808.
Crossref - Muccee F, Ejaz S. Whole genome shotgun sequencing of POPs degrading bacterial community dwelling tannery effluents and petrol contaminated soil. Microbiol Res. 2020;238:126504.
Crossref - Nipun B, Dholaria M, Tailor AJ. Metagenomic Exploration Microbial Diversity of Industrial Effluent Contaminated Soils of Silvassa , India. 2024;9(6):1232-1250.
Crossref - Auti AM, Narwade NP, Deshpande NM, Dhotre DP. Microbiome and imputed metagenome study of crude and refined petroleum-oil-contaminated soils: Potential for hydrocarbon degradation and plant-growth promotion. J Biosci. 2019;44(5):114.
Crossref - Gana M, Kure JT, Ahmadu U. The application of metagenomics in hydrocarbon resource management. Brazilian J Biol Sci. 2019;6(13):429-438.
Crossref - Hemmat-Jou MH, Safari-Sinegani AA, Mirzaie-Asl A, Tahmourespour A. Analysis of microbial communities in heavy metals-contaminated soils using the metagenomic approach. Ecotoxicology. 2018;27(9):1281-1291.
Crossref - Li YQ, Xin Y, Li C, Liu J, Huang T. Metagenomics-metabolomics analysis of microbial function and metabolism in petroleum-contaminated soil. Braz J Microbiol. 2023;54(2):935-947.
Crossref - Salam MD, Varma A. A review on impact of E-waste on soil microbial community and ecosystem function. Pollution. 2019;5(4):761-774.
Crossref - Junghare M, Spiteller D, Schink B. Anaerobic degradation of xenobiotic isophthalate by the fermenting bacterium Syntrophorhabdus aromaticivorans. ISME J. 2019;13(5):1252-1268.
Crossref - Medic A, Ljesevic M, Inui H, et al. Efficient biodegradation of petroleum: n-alkanes and polycyclic aromatic hydrocarbons by polyextremophilic Pseudomonas aeruginosa san ai with multidegradative capacity. RSC Adv. 2020;10(24):14060-14070.
Crossref - Boll M, Geiger R, Junghare M, Schink B. Microbial degradation of phthalates: biochemistry and environmental implications. Environ Microbiol Rep. 2020;12(1):3-15.
Crossref - Hidalgo KJ, Teramoto EH, Soriano AU, et al. Taxonomic and functional diversity of the microbiome in a jet fuel contaminated site as revealed by combined application of in situ microcosms with metagenomic analysis. Sci Total Environ. 2020;708:135152.
Crossref - Huang L, Ye J, Jiang K, Wang Y, Li Y. Oil contamination drives the transformation of soil microbial communities: Co-occurrence pattern, metabolic enzymes and culturable hydrocarbon-degrading bacteria. Ecotoxicol Environ Saf. 2021;225:112740.
Crossref - Bonomo MG, Calabrone L, Scrano L, et al. Metagenomic monitoring of soil bacterial community after the construction of a crude oil flowline. Environ Monit Assess. 2022;194(2).
Crossref - Ni J, Yang H, Chen L, et al. Metagenomic analysis of microbial community structure and function in a improved biofilter with odorous gases. Sci Rep. 2022;12(1):1731.
Crossref - Shah V, Zakrzewski M, Wibberg D, Eikmeyer F, Schlüter A, Madamwar D. Taxonomic Profiling and Metagenome Analysis of a Microbial Community from a Habitat Contaminated with Industrial Discharges. Microb Ecol. 2013;66(3):533-550.
Crossref - Luo Y, Zhou M, Zhao Q, et al. Complete genome sequence of Sphingomonas sp. Cra20, a drought resistant and plant growth promoting rhizobacteria. Genomics. 2020;112(5):3648-3657.
Crossref - Das N, Kotoky R, Maurya AP, Bhuyan B, Pandey P. Paradigm shift in antibiotic-resistome of petroleum hydrocarbon contaminated soil. Sci Total Environ. 2021;757:143777.
Crossref - Tiedje JM. Shewanella—the environmentally versatile genome. Nat Biotechnol. 2002;20(11):1093-1094.
Crossref - Mukherjee M, Zaiden N, Teng A, Hu Y, Cao B. Shewanella biofilm development and engineering for environmental and bioenergy applications. Curr Opin Chem Biol. 2020;59:84-92.
Crossref - Castrejon-Godinez ML, Ortiz-Hernandez ML, Salazar E, et al. Transcriptional analysis reveals the metabolic state of Burkholderia zhejiangensis CEIBS4-3 during methyl parathion degradation. PeerJ. 2019;7:e6822.
Crossref - Thomas JC, Oladeinde A, Kieran TJ, et al. Co-occurrence of antibiotic, biocide, and heavy metal resistance genes in bacteria from metal and radionuclide contaminated soils at the Savannah River Site. Microb Biotechnol. 2020;13(4):1179-1200.
Crossref - Wang M, Garrido-Sanz D, Sansegundo-Lobato P, et al. Soil Microbiome Structure and Function in Ecopiles Used to Remediate Petroleum-Contaminated Soil. Front Environ Sci. 2021;9:624070.
Crossref - Yan X, Jin W, Wu G, et al. Hydrolase CehA and Monooxygenase CfdC Are Responsible for Carbofuran Degradation in Sphingomonas sp. Strain CDS-1. Parales RE, ed. Appl Environ Microbiol. 2018;84(16):e00805-18.
Crossref - Zhang W, Lin Z, Pang S, Bhatt P, Chen S. Insights Into the Biodegradation of Lindane (g-Hexachlorocyclohexane) Using a Microbial System. Front Microbiol. 2020;11:522.
Crossref - Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017;45(D1):D353-D361.
Crossref - Kim JW, Hong YK, Kim HS, Oh EJ, Park YH, Kim SC. Metagenomic analysis for evaluating change in bacterial diversity in tph-contaminated soil after soil remediation. Toxics. 2021;9(12):319.
Crossref - Perez-Pantoja D, Donoso R, Junca H, Gonzalez B, Pieper DH. PPhylogenomics of Aerobic Bacterial Degradation of Aromatics. In: Timmis, K.N. (eds) Handbook of Hydrocarbon and Lipid Microbiology. Springer, Berlin, Heidelberg. 2010:1355-1397.
Crossref - Eze MO. Metagenome Analysis of a Hydrocarbon-Degrading Bacterial Consortium Reveals the Specific Roles of BTEX Biodegraders. Genes (Basel). 2021;12(1):98.
Crossref - Lee Y, Lee Y, Jeon CO. Biodegradation of naphthalene, BTEX, and aliphatic hydrocarbons by Paraburkholderia aromaticivorans BN5 isolated from petroleum-contaminated soil. Sci Rep. 2019;9(1):860.
Crossref - Silva CC, Hayden H, Sawbridge T, et al. Phylogenetic and functional diversity of metagenomic libraries of phenol degrading sludge from petroleum refinery wastewater treatment system. AMB Expr. 2012;2(1):1-13.
Crossref - Hernandez-Ospina DA, Osorio-Gonzalez CS, Miri S, Kaur Brar S. New perspectives on the anaerobic degradation of BTEX: Mechanisms, pathways, and intermediates. Chemosphere. 2024;361:142490.
Crossref - Wu HJ, Du XY, Wu WJ, Zheng J, Song JY, Xie JC. Metagenomic analysis reveals specific BTEX degrading microorganisms of a bacterial consortium. AMB Expr. 2023;13(1):48.
Crossref - Andreote FD, Jimenez DJ, Chaves D, et al. The Microbiome of Brazilian Mangrove Sediments as Revealed by Metagenomics. PLoS One. 2012;7(6):e38600.
Crossref - Yergeau E, Sanschagrin S, Beaumier D, Greer CW. Metagenomic Analysis of the Bioremediation of Diesel-Contaminated Canadian High Arctic Soils. PLoS One. 2012;7(1):e30058.
Crossref
© The Author(s) 2025. Open Access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License which permits unrestricted use, sharing, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.